



Regulation of DNA Double Strand Break Repair in Tumors

PD Dr. Wael Mansour, Dipl.Biochem.
Dr. Sabrina Köcher, Dipl.Biotech.
Dr. Christoph Oing, Onkologie-Arzt
Mia Peters, Onkologie-Ärztlin
Alexandra Zielinski, BTA
Stefanie Meien, MTA
Adriana Perugachi Heinsohn, MTA
Mohamed Elsesy, BSc. Pharm., PhD Stud.
Shaimaa Fahmi, BSc. Pharm., PhD Stud.
Ayham Moustafa, PhD Stud.
Goal
Our goal is to make transformative discoveries that will provide important new insights into DSB repair processes that maintain genome and cellular integrity. We also explore how such insights can be translated to better understanding and treating cancer. Currently we focus mainly on the role of DNA damage response and DSB repair in the development and progression of prostate cancer. Figure 1 shows our work concept
Specific topics:
1. DSB repair hierarchy and repair pathway choice
2. Defects of DDR and DSB repair in prostate cancer
3. Analysis of DDR and DSB repair in prostate cancer clinical materials
Topic 1 — DSB repair hierarchy / DSB repair pathway choice
DNA double strand breaks (DSBs) represent one of the major threats to genome integrity. Eukaryotic cells possess at least three pathways for DSB repair: non-homologous end joining (NHEJ), single-strand annealing (SSA) and homologous recombination (HR).
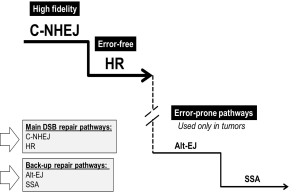
Figure 1 Representative functional hierarchy between DSB repair pathways. In normal cells, high fidelity NHEJ predominates over other repair pathways through Ku-mediated end protection. In the absence of Ku, the repair is switched to the error-prone PARP1-EJ and SSA. These error-prone pathways are frequently employed in tumor cells
DNA double strand breaks (DSBs) represent one of the major threats to genome integrity. Eukaryotic cells possess at least three pathways for DSB repair: non-homologous end joining (NHEJ), single-strand annealing (SSA) and homologous recombination (HR). Figure 1 Representative functional hierarchy between DSB repair pathways. In normal cells, high fidelity NHEJ predominates over other repair pathways through Ku-mediated end protection. In the absence of Ku, the repair is switched to the error-prone PARP1-EJ and SSA. These error-prone pathways are frequently employed in tumor cells These pathways differ not only in the underlying mechanism but also in their repair product. While HR is basically error-free repair pathway, NHEJ can be mutagenic and associated with some errors that might eventually lead to mutations. SSA on the other hand are the most mutagenic repair pathway which results in large deletion. In our lab we have identified a functional hierarchy between these repair pathways which ensures fast and faithful repair of the DSBs. According to this hierarchy, the accurate NHEJ (Classical-NHEJ; C-NHEJ) predominates over the other repair pathways. Consequently, when C-NHEJ is not an option cells switch the repair to the other DSB repair pathways (HR and SSA, Fig.1). Importantly, we could identify an alternative end joining pathway which predominates when C-NHEJ is defect. This pathway is slower, associated with long deletion length and more important PARP1-dependent because either inhibition or knockdown of PARP1 abrogates this pathway.
In addition we aim to specifically determine how cells choose the appropriate DSB repair pathway. The initial processing of the DSB ends is a key determinant of DSB repair pathway choice and is tightly regulated during the cell cycle. Both end protection factors (such as 53BP1, RIF1, and RAP80) and end resection factors (such as BRCA1, MRE11 and CtIP) directly influence the choice between NHEJ and HR by regulating 5’ end resection but how this is achieved remains unclear. Interestingly, all these factors are regulated by ataxia-telangiectasia mutated (ATM) gene which is the main coordinator of the DNA damage response after DSB. In the current project we aim to better understand the mechanism by which end protection and end resection factors cross-talk with each other to choose the appropriate repair pathway to be employed.
Topic-1-related publications
- Mansour WY, Schumacher S, Rosskopf R, Rhein T, Schmidt-Petersen F, Gatzemeier F, Haag F, Borgmann K, Willers H, Dahm-Daphi J (2008) Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res 36:4088-4098
- Kasten-Pisula U, Menegakis A, Brammer I, Borgmann K, Mansour WY, Degenhardt S, Krause M, Schreiber A, Dahm-Daphi J, Petersen C, Dikomey E, Baumann M (2009) The extreme radiosensitivity of the squamous cell carcinoma SKX is due to a defect in double-strand break repair. Radiother Oncol 90:257-264
- Mansour WY, Rhein T, Dahm-Daphi J (2010) The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res 38:6065-6077
- Kocher S, Rieckmann T, Rohaly G, Mansour WY, Dikomey E, Dornreiter I, Dahm-Daphi J (2012) Radiation-induced double-strand breaks require ATM but not Artemis for homologous recombination during S-phase. Nucleic Acids Res 40:8336-8347
- Mansour WY, Borgmann K, Petersen C, Dikomey E, Dahm-Daphi J (2013) The absence of Ku but not defects in classical non-homologous end-joining is required to trigger PARP1-dependent end-joining. DNA Repair (Amst) 12:1134-1142
- Mansour WY, Bogdanova NV, Kasten-Pisula U, Rieckmann T, Kocher S, Borgmann K, Baumann M, Krause M, Petersen C, Hu H, Gatti RA, Dikomey E, Dork T, Dahm-Daphi J (2013) Aberrant overexpression of miR-421 downregulates ATM and leads to a pronounced DSB repair defect and clinical hypersensitivity in SKX squamous cell carcinoma. Radiother Oncol 106:147-154
- A. Bakr, C. Oing, S. Köcher, K. Borgmann, I. Dornreiter, C. Petersen, E. Dikomey and W.Y. Mansour. "Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilamnet formation’’ Nucleic Acids Res. 2015 Mar 31;43(6):3154-66
- Bakr A, Köcher S, Volquardsen J, Reimer R, Borgmann K, Dikomey E, Rothkamm K, Mansour W.Y. ‘’Functional crosstalk between DNA damage response proteins 53BP1 and BRCA1 regulates double strand break repair choice’’. Radiother Oncol, 2016 May;119(2):276-81.
- A. Bakr, S. Köcher, J. Volquardsen, C. Petersen, K. Borgmann, E. Dikomey, K. Rothkamm and W. Y. Mansour. "Impaired 53BP1/RIF1 DSB-end protection stimulates CtIP-dependent end resection and switches the repair to PARP1-dependent end joining in G1". Oncotarget, 2016
Topic 2 — Impact of DSB repair in development and targeting of prostate cancer
Prostate cancer (PCa) is the most frequent cancer for men. Generally, PCa grows slowly and initially responds to androgen ablation therapy (androgen-dependent, AD). However, eventually some cases become refractory to this therapy, and progress into androgen-independent prostate cancers (Hormone refractory, HRPC). So far, little information is available about the molecular mechanisms underlying the development and progression of PCa. It is believed that progression of PCa obviously occurs via accumulating various DDR defects that lead to gross chromosomal aberrations such as translocations and deletions. Figure 2 shows our work concept for the (i) identification and exploitation of such defects, leading to better understanding of the molecular mechanism(s) of PCa progression and eventually identification of molecular biomarkers that can be used as predictors or modulators for the response of PCa to different treatment strategies.
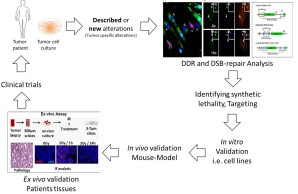
Figure 2 Schematic representation of AG2 work concept
Topic-2-related publications
- A. Kötter, K. Cornils, K. Borgmann, J.Dahm-Daphi, C. Petersen, E. Dikomey and W. Y. Mansour. "Inhibition of PARP1-ddependent end joining contributes to Olaparib-mediated radio-sensitization in tumor cells’’ Molecular Oncology, 2014.Mol Oncol. 2014, Dec; 8(8):1616-25.
- Kari V*, Mansour WY*, Raul SK, Baumgart SJ, Mund A, Grade M, Sirma H, Simon R, Will H, Dobbelstein M, Dikomey E, Johnsen SA. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016 Nov;17(11):1609-1623. * Equally contributed.
- C. Oing, Wael Y. Mansour, and C. Bokemeyer. "DNA-Reparaturdefekte und Olaparib". InfoOnkologie, 2016 (12-14).
- C. Oing, P. Tennstedt, R. Simon, J. Volquardsen, C. Bokemeyer, C. Petersen, E. Dikomey, K. Rothkamm, W. Y. Mansour. Bcl2 overexpressing cells are radiosensitized by PARP inhibitors due to repair switch to PARP1-dependent end joining, Cancer Letters, 2018, m423:60-70
- Mansour W.Y., Tennstedt P., Volquardsen J., Oing C., Kluth M., Hube-Magg C., Borgmann K., Simon R., Petersen C., Dikomey E., Rothkamm K. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Scientific Rep, 2018: 8:3947
- Köcher S, Beyer B, Lange T, et al. A functional ex vivo assay to detect PARP1-EJ repair and radiosensitization by PARP-inhibitor in prostate cancer. Int J Cancer. 2019;144(7):1685‐1696. doi:10.1002/ijc.32018
- M. Elsesy, S. J. Oh-Hohenhorst, A. Löser, C. Oing, S. Mutiara, S. Köcher, S. Meien, A. Zielinski, S. Burdak-Rothkamm, D. Tilki, H. Huland, R. Schwarz, C. Petersen, C. Bokemeyer, K. Rothkamm, W. Mansour. Second-generation antiandrogen therapy radiosensitizes prostate cancer regardless of castration state through inhibition of DNA double strand break repair, in press
Grants: BMBF, HH-KG, UCCH-Ein-Drittelstipendium Spierling-Stiftung, DAAD-GERLS
Topic 3- Analysis of DDR and DSB repair in prostate cancer clinical materials
We also successfully established an ex-vivo assay using tumor samples to follow DSB repair as well as to test new treatment strategies. This assay was already used to identify DSB repair defects in human xenograft tumors as well as in numerous human tumor samples. Using this assay we could establish an enhancement ration for the radiosensitization mediated by different drugs including olaparib and second generation antiandrogens.
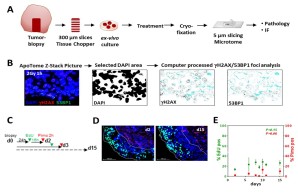
Figure 3 (A) Flow chart for ex-vivo assay steps. (B) Example for computer processed gH2AX/53BP1 foci analysis. (C) Flow chart for detection of proliferation (EdU) and hypoxia (Pimonidazol) for up to 15 days. (D) Representative micrographs for EdU (green) and Pimonidazol (red) positive cells. (E) Quantitation of EdU positive or Pimonidazol positive areas of the samples to control proliferation and hypoxia over 15 days. Adapted from Köcher et al., Int. J. Cancer, 2019
Topic-3-related publications
- Köcher S, Beyer B, Lange T, et al. A functional ex vivo assay to detect PARP1-EJ repair and radiosensitization by PARP-inhibitor in prostate cancer. Int J Cancer. 2019;144(7):1685‐1696. doi:10.1002/ijc.32018
Grants: BMBF, FFM, Monika Kutzner Stiftung
MSNZ Partner Lab (2020-)
In the context of Mildred-Scheel Cancer Career Center HATRICS4 , Dr. C. Oing and PD. Dr. W. Mansour applied successfully to get a partner lab together. The main aim of this we were successfully Dissemination of tumour cells and metastatic spread as well as cancer resistance to standard treatment are still the common cause of treatment failure and eventually death in prostate cancer patients.
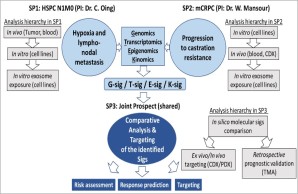
Figure 4 A schematic representation of the DRAGOON study
A better understanding the biology of the above-mentioned features will indeed lead to more effective cancer therapy options in prostate cancer. To that end, molecular techniques mixed with scientific experience of the medical scientist Dr Mansour complemented by clinical medical oncology expertise of Dr. Oing are gathered to implement the proposed bidirectional translational research project entitled DRAGOON. In DRAGOON (Figure 4), several high throughput techniques in various in vivo, in vitro and ex vivo models will be employed to address features of dissemination and metastasis as well as endocrine resistance in this context, by examining incurable prostate cancer at (i) an early (hormone-sensitive, node-positive) (subproject 1) and also (ii) a late (metastatic castration-resistant) (subproject 2) stage of metastatic disease as a special exemplar, taking into account the complexity of advanced prostate cancer. Both subprojects aim to identify distinct signatures within the genome, transcriptome, epigenome and/or kinome, which contribute to prostate cancer dissemination, metastasis and progression to castration resistant phenotype, which will assist in defining new biomarkers and novel, biomarker-driven, personalized therapeutic strategies (subproject 3).
Grants: Mildred-Scheel, German Cancer Aid