
- Forschungsgruppenleiter
Forschungsprofile
Ziele
Unser Hauptinteresse gilt der proteolytischen Verarbeitung bestimmter Schlüsselproteine (wie dem Prionprotein) und ihrer Bedeutung für Gesundheit und Krankheit, mit besonderem Schwerpunkt auf neurodegenerativen Erkrankungen. Zu letzteren gehören seltene und übertragbare Prionenkrankheiten (z. B. die Creutzfeldt-Jakob-Krankheit) und (viel) häufigere Krankheiten wie Alzheimer oder Parkinson. Alle diese schädlichen Krankheiten stellen Patienten, Angehörige, Pflegekräfte, das öffentliche Gesundheitswesen und die Gesellschaft als Ganzes vor enorme Leiden und Herausforderungen, und bis heute gibt es keine ursächlichen - und damit wirklich nützlichen - Behandlungen. Diese Krankheiten haben jedoch auch kritische mechanistische Prozesse gemeinsam, die zu einer Pathologie führen, die in Zukunft wahrscheinlich für gemeinsame therapeutische Ansätze relevant sein wird. Unsere Strategie besteht darin, evolutionär konservierte endogene enzymatische Spaltungen zu nutzen (d. h. die Fragmentierung von Proteinen durch "molekulare Scheren", die in allen unseren Körpern zu jeder Zeit stattfindet). Viele dieser Spaltungen und die daraus resultierenden Fragmente scheinen eine (neuro-)protektive Rolle zu spielen. Mit anderen Worten: Wir versuchen uns physiologische Prozesse zunutze zu machen, die von der Natur bereits vorgesehen sind. Zu diesem Zweck vertiefen wir ständig unser mechanistisches Wissen auf molekularer, zellulärer, geweblicher und organismischer Ebene, indem wir verschiedene Modellsysteme und Methoden verwenden. Darüber hinaus entwickeln wir neue Forschungsinstrumente (z. B. spaltstellenspezifische Antikörper), die kritische mechanistische Erkenntnisse ermöglichen und beschleunigen, die letztlich (und dringend) für neue therapeutische Perspektiven erforderlich sind. Unsere interdisziplinäre und translationale Ausrichtung wird auch durch eine enge und fruchtbare nationale und internationale Zusammenarbeit mit renommierten Experten auf den jeweiligen Gebieten erreicht.
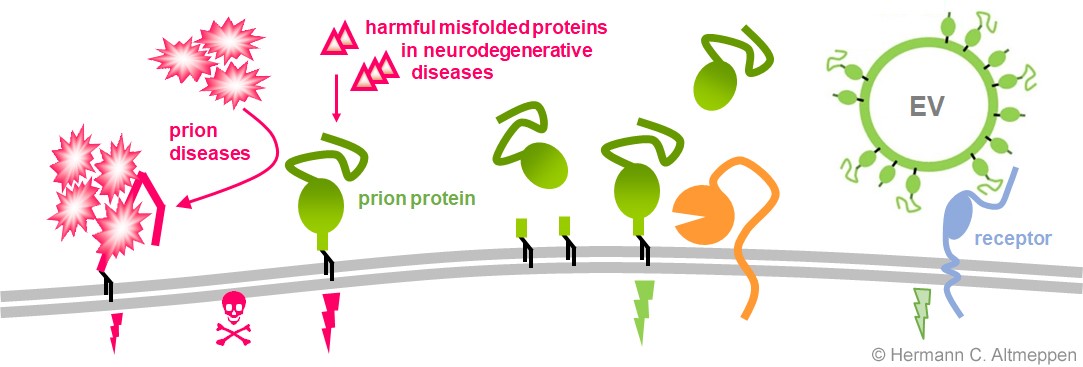
Das Prionprotein (grün) befindet sich auf der Oberfläche von Neuronen und vielen anderen Zelltypen, wo es seine physiologischen Funktionen ausübt (grüner Blitz). Bei Prionenkrankheiten (links) ist das Protein gezwungen, sich falsch zu falten (d. h. seine dreidimensionale Struktur zu verändern), wodurch es seine Funktionen verliert und beginnt, in einem fortschreitenden Prozess Aggregate zu bilden, die schließlich zum Tod führen. Das Prionprotein fungiert auch als zellulärer Rezeptor für andere toxische Proteine, die mit derzeit unheilbaren Gehirnkrankheiten wie Alzheimer oder Parkinson in Verbindung gebracht werden. Bestimmte Proteasen (oder "molekulare Scheren", orange) in unserem Körper spalten das Prionprotein ständig an bestimmten Stellen ab. Infolgedessen wird die Konzentration des Proteins an der Zelloberfläche verringert, und kürzere Fragmente des Proteins werden in den extrazellulären Raum freigesetzt, wo sie intrinsische biologische Funktionen haben und schützende Wirkungen entfalten können. Schließlich wird das Prionprotein auch in Verbindung mit extrazellulären Vesikeln (EV) aus den Zellen freigesetzt und könnte in diesen relevanten Strukturen eine wichtige Rolle bei der interzellulären Kommunikation in Gesundheit und Krankheit spielen.
Aktuelle Projekte
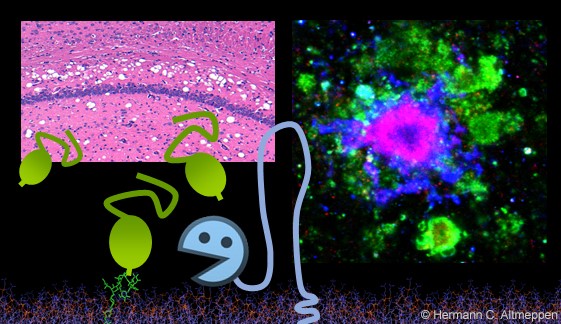
Die ADAM10-vermittelte Freisetzung des Prionproteins - Ein wenig aufgeben, um viel zu gewinnen?
Das Prionprotein (PrP) ist ein Hauptakteur bei neurodegenerativen Erkrankungen. Auf der Oberfläche von Nervenzellen fungiert es als Substrat für die schädliche und fortschreitende Fehlfaltung bei Prionenkrankheiten und als toxizitätsvermittelnder Rezeptor für schädliche Proteinkonformer bei anderen neurodegenerativen Krankheiten. Im Gegensatz dazu reduziert die Freisetzung von PrP durch die Metalloprotease ADAM10 sowie die Spaltung durch andere Enzyme den PrP-Spiegel an der Plasmamembran und erzeugt lösliche Fragmente, die bei neurodegenerativen Erkrankungen eine schützende Rolle zu spielen scheinen. Wir untersuchen die biologischen Auswirkungen dieser Spaltungen und der entstehenden Fragmente und wie diese für die Therapie genutzt werden könnten.
Ausgewählte Publikationen
- Ligands binding to the prion protein induce its proteolytic release with therapeutic potential in neurodegenerative proteinopathies.
Linsenmeier L§, Mohammadi B§, Shafiq M, Frontzek K, Bär J, Shrivastava A, Damme M., Schwarz A, Da Vela S, Massignan T, Jung S, Correia A, Schmitz M, Puig B, Hornemann S, Zerr I, Tatzelt J, Biasini E, Saftig P, Schweizer M, Svergun D, Amin L, Mazzola F, Varani L, Thapa S, Gilch S, Schätzl H, Harris D, Triller A, Mikhaylova M, Aguzzi A, Altmeppen HC§*, Glatzel M*. (in press) Science Advances [§equal contribution; *corresponding authors]
DOI: 10.1101/2021.04.19.440495 - GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice.
Puig B, Altmeppen HC et al. (2019) PLoS Pathogens
DOI: 10.1371/journal.ppat.1007520 - Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein.
Linsenmeier L, Mohammadi B, Wetzel S, Puig B, Jackson W, Hartmann A, Uchiyama K, Sakaguchi S, Endres K, Tatzelt J, Saftig P, Glatzel M*, Altmeppen HC*. (2018) Molecular Neurodegeneration [*corresponding authors]
DOI: 10.1186/s13024-018-0248-6 - The sheddase ADAM10 is a potent modulator of prion disease.
Altmeppen HC et al. (2015) eLife
DOI: 10.7554/eLife.04260 - Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo.
Altmeppen HC et al. (2011) Molecular Neurodegeneration
DOI: 10.1186/1750-1326-6-36

Andere Aspekte enzymatischer Spaltungen in der PrP-Biologie und/oder bei neurodegenerativen Erkrankungen
Zusätzlich zu den oben genannten Aspekten interessieren wir uns für die biologischen Funktionen des Prionproteins und seiner freigesetzten Formen. Außerdem untersuchen wir, ob der Nachweis spezifischer PrP-Fragmente etwas über Krankheitszustände bei Neurodegeneration, Krebs und anderen pathologischen Zuständen aussagen kann. Zu diesem Zweck untersuchen wir das Potenzial solcher Fragmente als aussagekräftige Biomarker für eine bessere und frühere molekulare Diagnose.
Gemeinsam mit unseren Mitarbeitern untersuchen wir auch die proteolytische Verarbeitung und die molekulare Beteiligung anderer Proteine, die kritisch mit Erkrankungen des Gehirns und darüber hinaus in Verbindung gebracht werden.
Ausgewählte Publikationen
- Transgenic Overexpression of the Disordered Prion Protein N1 Fragment in Mice Does Not Protect Against Neurodegenerative Diseases Due to Impaired ER Translocation.
Mohammadi B, Linsenmeier L, Shafiq M, Puig B, Galliciotti G, Giudici C, Willem M, Eden T, Koch-Nolte F, Lin YH, Tatzelt J, Glatzel M, Altmeppen HC. (2020) Molecular Neurobiology
DOI: 10.1007/s12035-020-01917-2 - Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease.
Sevillano AM, Aguilar-Calvo P, Kurt TD, Lawrence JA, Soldau K, Nam TH, Schumann T, Pizzo DP, Nyström S, Choudhury B, Altmeppen HC et al. (2020) Journal of Clinical Investigation
DOI: 10.1172/JCI131564 - In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15.
Seipold L, Altmeppen HC et al. (2018) Cellular and Molecular Life Science
DOI: 10.1007/s00018-018-2791-2 - Generation of aggregation prone N-terminally truncated amyloid β peptides by meprin β depends on the sequence specificity at the cleavage site.
Schönherr C, Schönherr C, Bien J, Isbert S, Wichert R, Prox J, Altmeppen HC et al. (2016) Molecular Neurodegeneration
DOI: 10.1186/s13024-016-0084-5 - High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer's disease.
Dohler F, Sepulveda-Falla D, Krasemann S, Altmeppen HC et al. (2014) Brain
DOI: 10.1093/brain/awt375
Das Prionprotein auf extrazellulären Vesikeln - funktionelle und krankheitsbezogene Aspekte
Das Prionprotein kann auch über extrazelluläre Vesikel (EV) freigesetzt werden. Letztere sind membranumhüllte Partikel, die von fast allen Zelltypen im Körper ausgeschieden werden. EVs werden zunehmend als wichtige Kommunikationsmittel zwischen Zellen erkannt und intensiv erforscht, die für eine Vielzahl von physiologischen und pathologischen Prozessen von Bedeutung sind. Sie stellen auch ein Mittel für zukünftige Therapien dar. Ein tieferes Verständnis der Schlüsselprozesse (z. B. Aufnahme und Verarbeitung von EVs durch Empfängerzellen) ist jedoch dringend erforderlich. Vor allem das Prionprotein ist auf EVs sehr häufig, wo es möglicherweise wichtige regulatorische Funktionen ausübt.
Ausgewählte Publikationen
- Brain-Derived Extracellular Vesicles in Health and Disease: A Methodological Perspective.
Brenna S, Krisp C, Altmeppen HC et al. (2021) International Journal of Molecular Sciences
DOI: 10.3390/ijms22031365 - Characterization of brain-derived extracellular vesicles reveals changes in cellular origin after stroke and enrichment of the prion protein with a potential role in cellular uptake.
Brenna S, Altmeppen HC et al. (2020) Journal of Extracellular Vesicles
DOI: 10.1080/20013078.2020.1809065 - Muskelin Coordinates PrPC Lysosome versus Exosome Targeting and Impacts Prion Disease Progression.
Heisler FF, Pechmann Y, Wieser I, Altmeppen HC et al. (2018) Neuron
DOI: 10.1016/j.neuron.2018.08.010 - Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein.
Linsenmeier L, Mohammadi B, Wetzel S, Puig B, Jackson WS, Hartmann A, Uchiyama K, Sakaguchi S, Endres K, Tatzelt J, Saftig P, Glatzel M*, Altmeppen HC*. (2018) Molecular Neurodegeneration [*corresponding authors]
DOI: 10.1186/s13024-018-0248-6 - Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity.
Falker C, Hartmann A, Guett I, Dohler F, Altmeppen HC et al. (2016) Journal of Neurochemistry
DOI: 10.1111/jnc.13514
Spenden
Wir danken der Creutzfeldt-Jakob Disease Foundation, Inc. ; der Alzheimer Forschung Initiative e.V. (AFI) und der Werner-Otto-Stiftung für die finanzielle Unterstützung unserer früheren und aktuellen Forschung.
Die Forschung befindet sich in einem ständigen Wandel. Die Lösung eines wissenschaftlichen Problems wirft mit Sicherheit mehrere neue Fragen und Herausforderungen auf. Wenn Sie unsere aktuellen und zukünftigen Projekte mit einer großzügigen Spende unterstützen möchten (unabhängig vom Betrag - jede Unterstützung hilft uns, unsere wissenschaftlichen Ziele zu erreichen), können Sie dies mit den folgenden Informationen, durch Scannen des QR-Codes oder durch einen Klick hier tun. Wir wissen Ihre Unterstützung sehr zu schätzen!

SPENDENKONTO
Verwendungszweck: 1345/001
Empfänger: UKE gemeinnützige GmbH
Bank: Hamburger Sparkasse
IBAN: DE 54 200 50 550 1234 363636
BIC: HASPDEHHXXX