Neuronal ceroid-lipofuscinosis type 7 (CLN7)
Project Leader: Stephan Storch
- Research
- Team
- Publications
- Funding
- Open positions
-
Research
CLN7 disease belongs to the group of neuronal ceroid lipofuscinoses (NCLs) which represent lysosomal storage disorders characterized by progressive photoreceptor- and neurodegeneration, neuroinflammation and lysosomal accumulation of autofluorescent ceroid lipopigments in affected children. All of the more than 30 known mutations in the CLN7/MFSD8 gene with two exceptions lead to CLN7 disease with variant late infantile phenotype. The CLN7/MFSD8 gene encodes a 518 aa polytopic lysosomal membrane protein with putative transporter function.
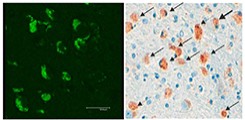
In our group we have generated the first mouse models for CLN7 disease ( Damme et al., 2014 ; Brandenstein et al., 2016 ). The Cln7 knockout mouse recapitulates key hallmarks of human CLN7 disease including a neurological phenotype, increased mortality, accumulation of autofluorescent material in the brain, lysosomal dysfunction, neuroinflammation, impaired macroautophagy, and neurodegeneration in the brain and the retina (Figure 1). One key interest of our studies is to analyze the role of CLN7 for lysosomal biogenesis and function by performing metabolomic, lipidomic, proteomic analyses, analyzing pathomechanisms, identification of potential substrates, and protein-protein interaction analyses. In cooperation with Prof. Bartsch (UKE Hamburg) we study the importance of Cln7 for the homeostasis of photoreceptors in the retina. The functional analyses include the heterologous expression of CLN7 at the plasma membrane and whole-cell uptake studies with radioactive substrates ( Steenhuis et al., 2012 ; Steenhuis et al., 2010 ). In collaboration with members of the BATCure consortium we will test experimental therapies aimed to reduce neurodegeneration and neuroinflammation in the retina and the brain of mutant mice.
-
-
Team
Project Leader
 Priv.-Doz. Dr. rer. nat.Stephan StorchPhoneE-mail
Priv.-Doz. Dr. rer. nat.Stephan StorchPhoneE-mailPostdoctoral Fellows
 Dr. rer. nat.Tatyana DanyukovaPhoneE-mail
Dr. rer. nat.Tatyana DanyukovaPhoneE-mailGraduate Students
Former Members
Sneha Nemani (M. Sc.)
Dr. rer. nat. Laura Brandenstein
Dr. rer. nat. Mine Franke
Dr. rer. nat. Pieter Steenhuis
Dr. med. Jana Galal
Dr. med. Adrian Meder
-
Publications
Selected Publications
- Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7. Brandenstein L, Schweizer M, Sedlacik J, Fiehler J, Storch S (2016) Hum Mol Genet 25:777-91 Abstract
- Gene disruption of Mfsd8 in mice provides the first animal model for CLN7 disease. Damme M, Brandenstein L, Fehr S, Jankowiak W, Bartsch U, Schweizer M, Hermans-Borgmeyer I, Storch S (2014) Neurobiol Dis 65:12-24 Abstract
- Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Kollmann K, Uusi-Rauva K, Scifo E, Tyynelä J, Jalanko A, Braulke T (2013) Biochim Biophys Acta 1832:1866-81 Abstract
- Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Kousi M, Lehesjoki AE, Mole SE (2012) Hum Mutat 33:42-63 Abstract
- Proteolytic cleavage of the disease-related lysosomal membrane glycoprotein CLN7. Steenhuis P, Froemming J, Reinheckel T Storch S (2012) Biochim Biophys Acta 1822:1617-28 Abstract
- Lysosomal targeting of the CLN7 membrane glycoprotein and transport via the plasma membrane require a dileucine motif. Steenhuis P, Herder S, Gelis S, Braulke T and Storch S (2010) Traffic 11:987-1000 Abstract
- The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Siintola E, Topcu M, Aula N, Lohi H, Minassian BA, Paterson AD, Liu XQ, Wilson C, Lahtinen U, Anttonen AK, and Lehesjoki AE (2007) Am J Hum Genet 81:136-46 Abstract
-
Funding
01/2016 - 12/2018
EU Horizon 2020
BATCure: European network for coordinated research on neuronal ceroid lipofuscinosis
Batten Disease Research Association (BDSRA)
Website BDSRA
5/2008 - 4/2017
German Research Foundation (DFG)
Research Training Group 1459:
"Sorting and Interactions between Proteins of Subcellular Compartments"
www.grk1459.de
09/2012 - 08/2015
German Research Foundation (DFG)
Grant STO761/3-1
„Erzeugung und Analyse eines Mausmodells der
variant spätinfantilen neuronalen Ceroid-Lipofuszinose“
http://gepris.dfg.de/gepris/projekt/224206679 -
-
Open positions
The lab is always interested in applications of highly motivated PhD, Bachelor and Master students who are interested in exciting molecular aspects of lysosomal cell biology and neurodegeneration. Qualified students are encouraged to submit their complete applications to:
PD Dr. Stephan Storch
UKE Children's hospital
Section Biochemistry
Martinistr. 46
D-20246 Hamburgemail: storch@uke.de