Our Research Focus
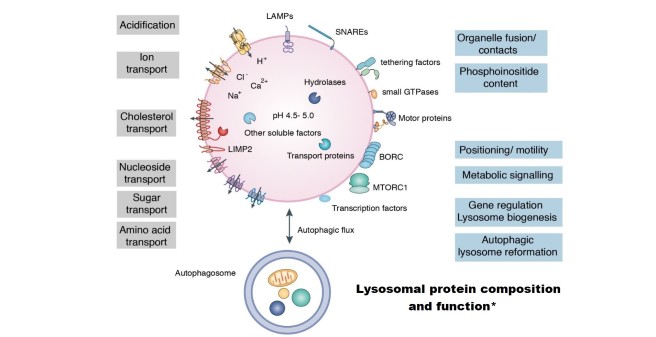
Lysosomes control the degradation, processing and recycling of proteins, lipids, glycans and nucleic acids delivered to the organelles through the biosynthetic, endocytic, phagocytic or autophagic routes. These processes are catalyzed and mediated by ~70 hydrolytic enzymes, accessory proteins and more than 250 membrane proteins, transporters and ion channels. Lysosomes can also fuse with autophagosomes to recycle intracellular nutrients and eliminate protein aggregates, damaged mitochondria and lysosomes as well as bacteria.
In recent years many proteins have been identified on the cytosolic surface of lysosomes that expand the functions of lysosomes mediating e.g. fusogenic and non-fusogenic contacts with other organelles, lysosomal positioning and motility, or metabolic sensing and gene regulation.
Numerous inherited defects in lysosomal enzymes result in lysosomal dysfunction, accumulation of undigested substrates and alterations in the size, ultrastructure, and cellular positioning of lysosomes. We investigate
· The role of mannose 6-phosphate (M6P) modifications on lysosomal enzymes for their sorting and function, and the M6P-independent transport routes to lysosomes, and
· The transcriptional regulation of lysosomal biogenesis.
To study these topics, we apply state-of-the-art cell biological methods, imaging techniques and biochemical approaches, including quantitative mass spectrometry, to analyze cellular, organoid and mouse models of human diseases. The advanced knowledge from this research will further our understanding i) of lysosomes in the maintenance of cell or organismal homeostasis, ii) pathomechanisms causing lysosome-related diseases such as mucolipidosis II and III and neuronal ceroid lipofuscinoses, and iii) for the development of novel therapeutic strategies.
*figure modified from Ballabio & Bonifacino (Nat Rev Mol Cell Biol. 2020; 21:101-118)