Pathophysiology of Glutaric Aciduria Type 1
- Research
- Team
- Diagnostics and Cooperations
- Publications
- Funding
- Links
-
Research
The inherited neurometabolic disorder glutaric aciduria type 1 (GA1) results from mutations in the gene for the mitochondrial matrix enzyme glutaryl-CoA dehydrogenase (GCDH). GCDH catalyzes the oxidative decarboxylation of glutaryl-CoA in the degradative pathway of the amino acids lysine, hydroxylysine and tryptophan (see Figure).
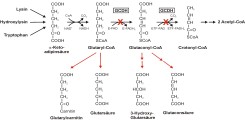

Figure: Degradative pathway of lysine, hydroxylysine and tryptophan. GCDH mediates the oxidative decarboxylation of glutaryl-CoA to crotonyl-CoA in two steps. In GCDH deficiency, glutaryl-CoA breaks down to glutarylcarnitine and glutaric acid, whereas glutaconyl-CoA is converted to 3-hydroxyglutaric acid and glutaconate by yet unknown mechanisms.
Affected patients are at risk for the development of encephalopathic crises triggered by catabolic situations such as infectious diseases, fever or diarrhea during a time window from birth to 36 months of age. An encephalopathic crisis is accompanied by a further increase of GA and 3OHGA concentrations and a subsequent, selective destruction of striatal neurons (Fig. 2) followed by an irreversible disabling movement disorder. The underlying pathophysiologic mechanisms are poorly understood. The clinical course of GA1 shows a high degree of variability, and some patients remain asymptomatic (Heringer et al. 2010). Considerable variation in severity of the clinical phenotype is observed with no correlation to the genotype.
The aim of our research is to investigate
- the underlying pathophysiologic mechanisms contributing to neurodegeneration in a GA1 mouse model,
- the coordinated action of different transporter proteins translocating the charged dicarboxylate metabolites across various membranes,
- functional and biochemical effects of specific mutations of the GCDH protein on the cellular level.
-
-
Team
Scientific Staff
Ann-Cathrin Dörfler, Medical student
Benjamin Lohmöller, Medical student
-
Diagnostics and Cooperations
Routine Diagnostics
Genetic analysis of the GCDH-coding gene is performed in the Laboratory for Genetics of Metabolic Diseases, Prof. R. Santer. For information please contact muehlhausen@uke.de.Cooperations
- Prof. B. C. Burckhardt und Prof. G. Burckhardt, Institut für Vegetative Physiologie und Pathophysiologie, Georg-August-Universität Göttingen
- Prof. E. Christensen, Dept. of Clinical Genetics, Rigshospitalet, Kopenhagen, Dänemark
- Prof. S. I. Goodman, Dr. M. Woontner, University of Colorado, Denver, USA
- Prof. D. Koeller, Oregon Health and Science University, Portland, USA
- Prof. Dr. S. Kölker, Universitäts-Kinderklinik, Heidelberg
- Dr. Z. Lukacs, Pädiatrisches Stoffwechsellabor, Universitätsklinikum Hamburg-Eppendorf
-
Publications
Project-relevant Publications
Original articles:
- Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, Ro J, Wagner GR, Green MF, Madsen AS, Schmiesing J, Peterson BS, Xu G, Ilkayeva OR, Muehlbauer MJ, Braulke T, Mühlhausen C, Backos DS, Olsen CA, McGuire PJ, Pletcher SD, Lombard DB, Hirschey MD, Zhao Y (2014) Cell Metab 19:605-17 Abstract
- Interaction of glutaric aciduria type 1-related glutaryl-CoA dehydrogenase with mitochondrial matrix proteins. Schmiesing J, Schlüter H, Ullrich K, Braulke T, Mühlhausen C (2014) PLoS One 9:e87715 Abstract
- Acute renal proximal tubule alterations during induced metabolic crises in a mouse model of glutaric aciduria type 1. Thies B, Meyer-Schwesinger C, Lamp J, Schweizer M, Koeller DM, Ullrich K, Braulke T, Mühlhausen C (2013) Biochim Biophys Acta 1832:1463-72 Abstract
- Glutaric aciduria type 1 metabolites impair the succinate transport from astrocytic to neuronal cells. Lamp J, Keyser B, Koeller DM, Ullrich K, Braulke T, Mühlhausen C (2011) J Biol Chem 286:17777-84 Abstract
- Disease-causing missense mutations affect enzymatic activity, stability and oligomerization of glutaryl-CoA dehydrogenase (GCDH). Keyser B, Mühlhausen C, Dickmanns A, Christensen E, Muschol N, Ullrich K, Braulke T (2008) Hum Mol Genet 17:3854-63 Abstract
- Transport and distribution of 3-hydroxyglutaric acid before and during induced encephalopathic crises in a mouse model of glutaric aciduria type 1. Keyser B, Glatzel M, Stellmer F, Kortmann B, Lukacs Z, Kölker S, Sauer SW, Muschol N, Herdering W, Thiem J, Goodman SI, Koeller DM, Ullrich K, Braulke T, Mühlhausen C (2008) Biochim Biophys Acta 1782:385-90 Abstract
- 3-Hydroxyglutaric acid is transported via the sodium-dependent dicarboxylate transporter NaDC3. Stellmer F, Keyser B, Burckhardt BC, Koepsell H, Streichert S, Glatzel M, Jabs S, Thiem J, Herdering W, Koeller DM, Goodman SI, Lukacs Z, Ullrich K, Burckhardt G, Braulke T, Mühlhausen C (2007) J Mol Med 85:763-70 Abstract
Reviews on Pathophysiology:
- The unsolved puzzle of neuropathogenesis in glutaric aciduria type I. Jafari P, Braissant O, Bonafé L, Ballhausen D (2011) Mol Genet Metab 104:425-37 Abstract
- Membrane translocation of glutaric acid and its derivatives. Mühlhausen C, Burckhardt BC, Hagos Y, Burckhardt G, Keyser B, Lukacs Z, Ullrich K, Braulke T (2008) J Inherit Metab Dis 31:188-93 Abstract
Therapy:
- Diagnosis and management of glutaric aciduria type I--revised recommendations. Kölker S, Christensen E, Leonard JV, Greenberg CR, Boneh A, Burlina AB, Burlina AP, Dixon M, Duran M, García Cazorla A, Goodman SI, Koeller DM, Kyllerman M, Mühlhausen C, Müller E, Okun JG, Wilcken B, Hoffmann GF, Burgard P (2011) J Inherit Metab Dis 34:677-694 Abstract
-
Funding

07/2014 - 06/2017
Chris Mühlhausen
German Research Foundation (DFG)
Grant MU 1778/3-2
„Analyse des Glutaryl-CoA-Dehydrogenase-
Multiprotein-Komplexes“
http://gepris.dfg.de/gepris/projekt/211332540 -
-
Links