
- Head of medical management
- Director of the institute
- Medical Specialist in Neuropathology
Research Profiles
Goals
We are especially interested in the mechanisms of synaptic dysfunction in prion diseases such as Creutzfeldt-Jakob disease. Additionally, we investigate the role the prion protein plays in dementias.
Generation and accumulation of abnormally processed proteins such as a misfolded form of the prion protein for Creutzfeldt-Jakob disease and beta-amyloid for Alzheimers disease lead to synaptic failure leading to irreversible nerve cell loss and progressive dementia. How the generation of pathological prions and deposition of these in the brain lead to neurodegeneration is not fully understood. Likewise, the role of the prion protein in Alzheimers disease or other dementias is not well defined.
Professor Glatzels’ lab addresses these questions experimentally by using a translational approach comparing data which are obtained by analyzing mouse models to data of patients suffering from Creutzfeldt-Jakob disease, Alzheimers disease or other dementias. For this, we employ a wide spectrum of techniques from molecular biology, biochemistry, and histochemistry. The Lab applies and further develops high-end imaging techniques and links –Omics analyses to clinical patient outcomes.
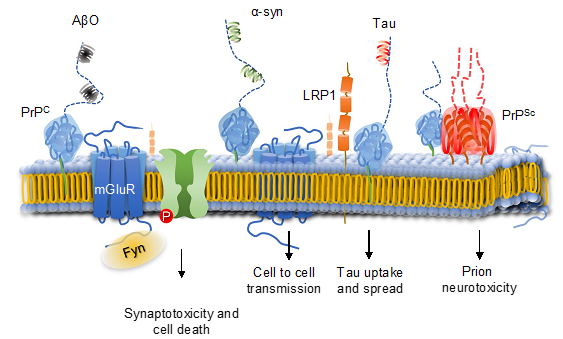
Overview of the multiple roles of the prion protein in Creutzfeldt-Jakob disease, Alzheimers disease and other dementias. The prion protein serves as a co-receptor for neurotoxic oligomeric species such as beta-amyloid and α-synuclein and mediates neurotoxicity via the Src-kinase Fyn. The prion protein is involved in the spread of Tau oligomers via LRP. The prion protein is also involved in transducing neurotoxicity caused by pathological prions (PrPSc).