
Medical Systems Biology
At the Institute of Medical Systems Biology (IMSB) we are dedicated to unraveling the complexities of human pathology, by combining expertise in AI, machine learning, bioinformatics, and image analysis.
Currently, we are focusing on understanding cell to cell signaling in immune mediated and other diseases.

“We enhance understanding of human pathology with computational methods, improving patient outcomes through innovative systems.”
Prof. Dr. Stefan Bonn
Project details and goals
We strive to deepen the understanding of human pathology through computational methodologies and ultimately improve patient outcomes through innovative clinical decision support systems and novel therapeutic approaches. We maintain close collaboration with the medical clinics of the UKE to ensure our research remains directly applicable to clinical practice. Our approach integrates and analyzes vast amounts of biomedical data using cutting-edge technologies such as multi-omics analyses, drug-target interaction, deep learning or spatial transcriptomics.
Our commitment to advancing scientific knowledge extends beyond our research efforts. We offer courses in machine and deep learning to foster interdisciplinary collaboration among physicists, computer scientists, and medical professionals. Additionally, our research groups focus on specialized areas such as genomic AI, computational pathology, biomedical data analysis, and data integration.
Current projects
MAXOMOD
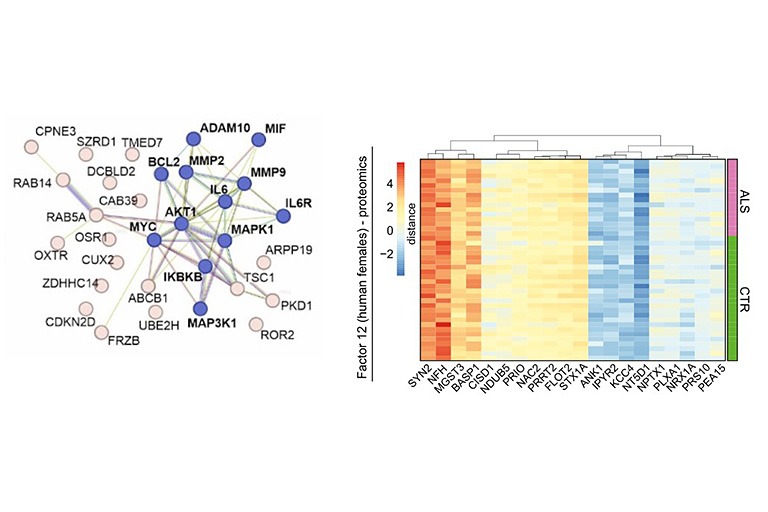
Motor neuron diseases such as amyotrophic lateral sclerosis (ALS) are rare neurodegenerative diseases with poor prognosis and insufficient treatment options. Patients with ALS develop increasing muscle weakness as the disease progresses, ultimately leading to death when lung function is lost. Existing therapy options only treat the symptoms of the disease and can hardly influence the course of neurodegeneration. Therefore, there is a need for research to ensure that new therapies arrive in the treatment of motor neuron disease. The goal of the project is to develop new therapeutic strategies as well as biomarkers that enable early diagnosis of the disease. To this end, new molecular targets are to be identified that could serve as potential targets for drug therapies.
Since RNA metabolism plays a crucial role in the pathogenesis of ALS, a protein-based approach will not be sufficient to comprehensively characterize disease-relevant pathways. We will thus combine multiple analytic methods from genomics, transcriptomics and microRNAomics up to proteomics, phosphoproteomics and metabolomics followed by multivariate semantic data integration to identify new disease-relevant pathways and biomarkers related to axono-synaptic pathology.
PCAI
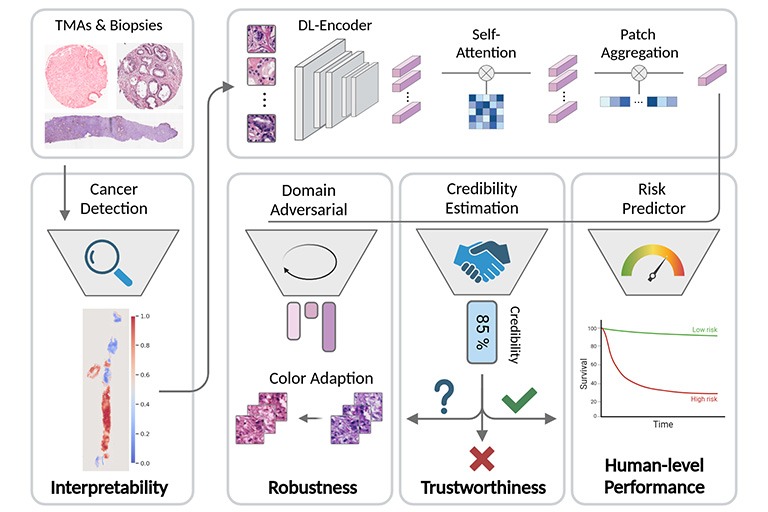
In clinical practice, the diagnosis of Prostate cancer (PCa) and the treatment decision are based on Gleason scores and nomograms. However, Gleason scores suffer from high inter-observer variability and nomograms are only able to model linear dependencies between patient parameters. Therefore, the goal of our research is to provide individual and objective prognoses for PCa patients through predicting relapse after radical prostatectomy (RPE). To this end we build and train deep learning-based models that predict the probability of a patient having a relapse from either H&E-stained images of biopsies after the RPE or electronic health records that reflect the patient’s clinical history related to prostate cancer.
DISCERN
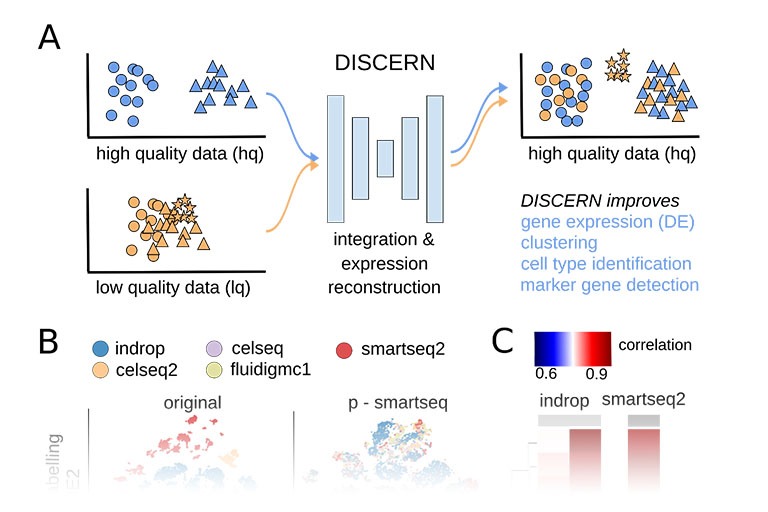
Single cell sequencing provides detailed insights into biological processes including cell differentiation and identity. While providing deep cell-specific information, the method suffers from technical constraints, most notably a limited number of expressed genes per cell, which leads to suboptimal clustering and cell type identification. We developed DISCERN, a novel deep generative neural network that reconstructs missing single cell gene expression using a reference dataset. DISCERN based expression inference results in greatly improved cell clustering, cell type and activity detection, and can lead insights into the cellular regulation of disease. For example, we used DISCERN to detect two cell types with a potential role in adverse COVID-19 outcome. DISCERN can be easily integrated into existing single cell sequencing workflows and readily adapted to enhance various other biomedical data types.
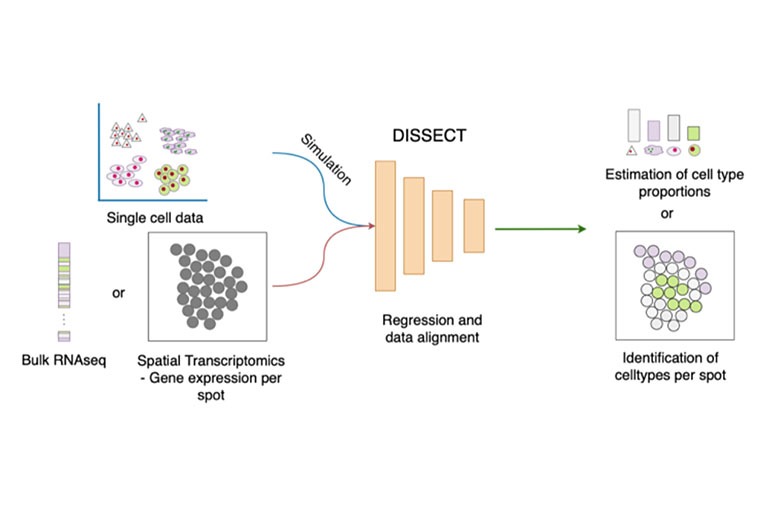
DISSECT
Bulk RNA-seq has enabled contributions in biomarker discovery, survival prediction and optimizing potential therapeutic targets. In comparison to single cell RNA-seq, the method is affordable and as such larger cohorts can be studied. While providing unprecedented insights at tissue level, the method is unsuitable to study cell type specific changes. Previously, IMSB developed SCADEN, a novel deep neural network ensemble framework that enabled accurate transfer of cell type specific knowledge from single cell RNA-seq to recover cell type landscape of bulk RNA-seq. For example, SCADEN revealed an increasing vascularization in Alzheimer’s disease. However single cell datasets suffer from dropouts and they differ in their scope from one study to another. Therefore, we developed DISSECT that utilizes information from bulk RNA-seq directly to estimate more accurate and robust cell type proportions. For example, we used DISSECT to recover cell type proportions of over 20 immune cell types in colons from individuals with inflammatory bowel disease where we identified an increasing infiltration of immune cells as well as a significantly increased number of activated resident cells. DISSECT is highly scalable and is easily integratable in existing bioinformatics workflows.
Publications 2025
Kuehl, M., Okabayashi, Y., Wong, M.N. et al. Nature 644, 516–526 (2025).
Abstract
The expression and location of proteins in tissues represent key determinants of health and disease. Although recent advances in multiplexed imaging have expanded the number of spatially accessible proteins, the integration of biological layers (that is, cell structure, subcellular domains and signalling activity) remains challenging. This is due to limitations in the compositions of antibody panels and image resolution, which together restrict the scope of image analysis. Here we present pathology-oriented multiplexing (PathoPlex), a scalable, quality-controlled and interpretable framework. It combines highly multiplexed imaging at subcellular resolution with a software package to extract and interpret protein co-expression patterns (clusters) across biological layers. PathoPlex was optimized to map more than 140 commercial antibodies at 80 nm per pixel across 95 iterative imaging cycles and provides pragmatic solutions to enable the simultaneous processing of at least 40 archival biopsy specimens. In a proof-of-concept experiment, we identified epithelial JUN activity as a key switch in immune-mediated kidney disease, thereby demonstrating that clusters can capture relevant pathological features. PathoPlex was then used to analyse human diabetic kidney disease. The framework linked patient-level clusters to organ disfunction and identified disease traits with therapeutic potential (that is, calcium-mediated tubular stress). Finally, PathoPlex was used to reveal renal stress-related clusters in individuals with type 2 diabetes without histological kidney disease. Moreover, tissue-based readouts were generated to assess responses to inhibitors of the glucose cotransporter SGLT2. In summary, PathoPlex paves the way towards democratizing multiplexed imaging and establishing integrative image analysis tools in complex tissues to support the development of next-generation pathology atlases.
Wang, H., Engesser, J., Khatri, R. et al. Nat Commun 16, 4686 (2025).
Abstract
In anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV) and systemic lupus erythematosus (SLE), glomerulonephritis is a severe kidney complication driven by immune cells, including T cells. However, the mechanisms underlying T cell activation in these contexts remain elusive. Here we report that in patients with AAV and SLE, type I interferon (IFN-I) induces T cell differentiation into interferon-stimulated genes-expressing T (ISG-T) cells, which are characterized by an elevated IFN-I signature, an immature phenotype, and cytotoxicity in inflamed tissue. Mechanistically, IFN-I stimulates the expression of interferon regulatory factor 7 (IRF7) in T cells, which in turn induces granzyme B production. In mice, blocking IFN-I signaling reduces IRF7 and granzyme B expression in T cells, thus ameliorating glomerulonephritis. In parallel, spatial transcriptomic analyses of kidney biopsies from patients with AAV or SLE reveal an elevated ISG signature and the presence of ISG-T cells in close proximity to plasmacytoid dendritic cells, the primary producers of IFN-I. Our results from both patients and animal models thus suggest that IFN-I production in inflamed tissue may drive ISG-T cell differentiation to expand the pool of cytotoxic T cells in autoimmune diseases.
Publications 2024
Engesser, J., Khatri, R., Schaub, D.P. et al. Nat Commun 15, 8220 (2024).
Abstract
Antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis is a life-threatening autoimmune disease that often results in kidney failure caused by crescentic glomerulonephritis (GN). To date, treatment of most patients with ANCA-GN relies on non-specific immunosuppressive agents, which may have serious adverse effects and be only partially effective. Here, using spatial and single-cell transcriptome analysis, we characterize inflammatory niches in kidney samples from 34 patients with ANCA-GN and identify proinflammatory, cytokine-producing CD4+ and CD8+ T cells as a pathogenic signature. We then utilize these transcriptomic profiles for digital pharmacology and identify ustekinumab, a monoclonal antibody targeting IL-12 and IL-23, as the strongest therapeutic drug to use. Moreover, four patients with relapsing ANCA-GN are treated with ustekinumab in combination with low-dose cyclophosphamide and steroids, with ustekinumab given subcutaneously (90 mg) at weeks 0, 4, 12, and 24. Patients are followed up for 26 weeks to find this treatment well-tolerated and inducing clinical responses, including improved kidney function and Birmingham Vasculitis Activity Score, in all ANCA-GN patients. Our findings thus suggest that targeting of pathogenic T cells in ANCA-GN patients with ustekinumab might represent a potential approach and warrants further investigation in clinical trials.
Previous publications
Hausmann, F., Ergen, C., Khatri, R., Marouf, M., Hänzelmann, S., Gagliani, N., ... & Bonn, S. (2023). Genome Biology, 24(1), 212.
Abstract
Background: Single-cell sequencing provides detailed insights into biological processes including cell differentiation and identity. While providing deep cell-specific information, the method suffers from technical constraints, most notably a limited number of expressed genes per cell, which leads to suboptimal clustering and cell type identification.
Results: Here, we present DISCERN, a novel deep generative network that precisely reconstructs missing single-cell gene expression using a reference dataset. DISCERN outperforms competing algorithms in expression inference resulting in greatly improved cell clustering, cell type and activity detection, and insights into the cellular regulation of disease. We show that DISCERN is robust against differences between batches and is able to keep biological differences between batches, which is a common problem for imputation and batch correction algorithms. We use DISCERN to detect two unseen COVID-19-associated T cell types, cytotoxic CD4+ and CD8+ Tc2 T helper cells, with a potential role in adverse disease outcome. We utilize T cell fraction information of patient blood to classify mild or severe COVID-19 with an AUROC of 80% that can serve as a biomarker of disease stage. DISCERN can be easily integrated into existing single-cell sequencing workflow.
Conclusions: Thus, DISCERN is a flexible tool for reconstructing missing single-cell gene expression using a reference dataset and can easily be applied to a variety of data sets yielding novel insights, e.g., into disease mechanisms.
Marouf, M., Machart, P., Bansal, V., Kilian, C., Magruder, D. S., Krebs, C. F., & Bonn, S. (2020). Nature communications, 11(1), 166.
Abstract
A fundamental problem in biomedical research is the low number of observations available, mostly due to a lack of available biosamples, prohibitive costs, or ethical reasons. Augmenting few real observations with generated in silico samples could lead to more robust analysis results and a higher reproducibility rate. Here, we propose the use of conditional single-cell generative adversarial neural networks (cscGAN) for the realistic generation of single-cell RNA-seq data. cscGAN learns non-linear gene–gene dependencies from complex, multiple cell type samples and uses this information to generate realistic cells of defined types. Augmenting sparse cell populations with cscGAN generated cells improves downstream analyses such as the detection of marker genes, the robustness and reliability of classifiers, the assessment of novel analysis algorithms, and might reduce the number of animal experiments and costs in consequence. cscGAN outperforms existing methods for single-cell RNA-seq data generation in quality and hold great promise for the realistic generation and augmentation of other biomedical data types.
Menden, K., Marouf, M., Oller, S., Dalmia, A., Magruder, D. S., Kloiber, K., ... & Bonn, S. (2020). Science advances, 6(30), eaba2619.
Abstract
A fundamental problem in biomedical research is the low number of observations available, mostly due to a lack of available biosamples, prohibitive costs, or ethical reasons. Augmenting few real observations with generated in silico samples could lead to more robust analysis results and a higher reproducibility rate. Here, we propose the use of conditional single-cell generative adversarial neural networks (cscGAN) for the realistic generation of single-cell RNA-seq data. cscGAN learns non-linear gene–gene dependencies from complex, multiple cell type samples and uses this information to generate realistic cells of defined types. Augmenting sparse cell populations with cscGAN generated cells improves downstream analyses such as the detection of marker genes, the robustness and reliability of classifiers, the assessment of novel analysis algorithms, and might reduce the number of animal experiments and costs in consequence. cscGAN outperforms existing methods for single-cell RNA-seq data generation in quality and hold great promise for the realistic generation and augmentation of other biomedical data types.
Halder, R., Hennion, M., Vidal, R. O., Shomroni, O., Rahman, R. U., Rajput, A., ... & Bonn, S. (2016). Nature neuroscience, 19(1), 102-110.
Abstract
The ability to form memories is a prerequisite for an organism's behavioral adaptation to environmental changes. At the molecular level, the acquisition and maintenance of memory requires changes in chromatin modifications. In an effort to unravel the epigenetic network underlying both short- and long-term memory, we examined chromatin modification changes in two distinct mouse brain regions, two cell types and three time points before and after contextual learning. We found that histone modifications predominantly changed during memory acquisition and correlated surprisingly little with changes in gene expression. Although long-lasting changes were almost exclusive to neurons, learning-related histone modification and DNA methylation changes also occurred in non-neuronal cell types, suggesting a functional role for non-neuronal cells in epigenetic learning. Finally, our data provide evidence for a molecular framework of memory acquisition and maintenance, wherein DNA methylation could alter the expression and splicing of genes involved in functional plasticity and synaptic wiring.
Team


Prof. Dr. med. Stefan Bonn
Head of lab - Systems Biology
E-mail address:















