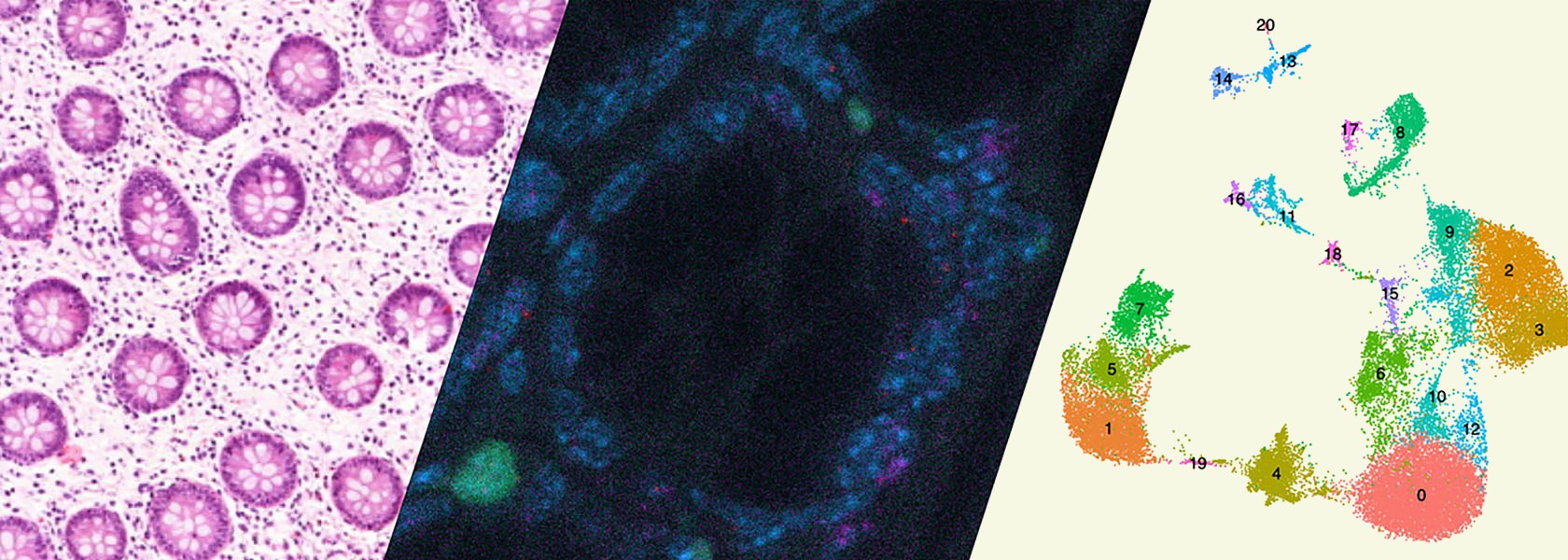

Intestinal immune regulation
The intestine plays a key role in controlling the immune response. By understanding the interplay between the microbiota, the tissue and the immune cells, we can pave the way for future therapies for chronic inflammatory diseases and cancer.

“The intestine plays a key role in controlling the immune response. By understanding the interplay between the microbiota, the tissue and the immune cells, we can pave the way for future therapies for chronic inflammatory diseases and cancer.”
Prof. Dr. Samuel Huber
Head of Molecular Gastroenterology and Immunology
Project details and goals
Inflammation is fundamental to promote tissue regeneration upon injury, and, in turn, the resolution of the immune response. Physiological tissue regeneration depends on fine-tuned interactions between the immune system, the tissue, and the microbiota. However, the complex communication between these three components and the molecules that mediate it are unclear. We hypothesize that inflammatory bowel disease (IBD) and colorectal cancer (CRC) are a consequence of a miscommunication between these components. Furthermore, this regulation also affects diseases in extra-intestinal organs. Thus, we hypothesize that by controlling the intestinal immune system we can prevent or cure chronic inflammatory diseases and even cancer also in extra-intestinal organs, such as the liver and kidney.
This hypothesis is based on recent findings by others and by us showing that the intestine plays a key role in modulating the balance between regulatory T cells, such as Foxp3+ Treg, Tr1 cells, and effector T cells, thereby influencing Immune-mediated inflammatory diseases in several organs. However, most T cells and cytokines, such as IL-22, have both context dependent pathogenic and protective functions. The right balance must be achieved. Thus, one key aspect of our study is to dissect complex biological systems in order to regulate rather than to block immune responses in order to facilitate the beneficial functions of these cells and cytokines.
Current projects
Project 1: IL-22 – IL-22BP: Spatio-Temporal Regulation of Inflammation and Tissue Regeneration
Interleukin-22 (IL-22) is one key orchestrator of this communication: It is produced by immune cells and by acting on intestinal epithelial cells, it modulates the composition of the microbiota and promotes tissue regeneration. However, IL-22 can also promote both chronic inflammation and cancer. Exactly what regulates these paradoxical effects remains unclear. Of note, there is an endogenous inhibitor of IL-22, namely IL-22 binding protein (IL-22BP), which blocks IL-22 activity. We hypothesize that a misguided spatio-temporal regulation of the IL-22 – IL-22BP axis is the cause of pathogenic effects of IL-22.
In particular, we will analyse: (i) the location, and the functional and molecular heterogeneity; (ii) the origin and fate of IL-22 and IL-22BP producing immune cells; and (iii) the role of the microbiota in regulating them. To this end, we will use new transgenic and gnotobiotic mouse models, single cell RNA sequencing and human samples.
In short, by studying the IL-22 - IL-22BP axis, we will understand how the complex interactions between the immune system, the tissue, and the microbiota lead to either physiological or pathological tissue regeneration. This will provide the basis for therapies controlling inflammation and tissue regeneration in a spatio-temporal manner.
Influence of environment factors on CD4+ T-helper cell plasticity and function
We aim to understand how different factors influence the phenotype and function of immune cells and their cytokine products. We are particularly focused on Inflammatory Bowel Diseases (IBD) and Colorectal Cancer (CRC). Among different factors, our focus is on the intestinal microbiota, dietary components such as gluten, and environmental plastics present in everyday products. By combining high-throughput techniques such as single-cell sequencing and microbiota sequencing with supervised techniques such as flow cytometry and qPCR, we analyze human samples collected from individuals with IBD before and after various biological treatments and dietary interventions. These diseases are also studied using animal models, e.g. with different but defined microbiota: SPF microbiota, ‘Wildling’ mice (with naturally occurring pathogens) as well as patient-specific gnotobiotic mice. We are furthermore interested in how the intestine can shape immune responses in other organs (such as the liver) and systemically. One of our particular focuses is understanding the role of individual T cells and cytokines in controlling the response to immune-checkpoint inhibitor treatment (both in terms of the oncological response and the development of side effects – immune related adverse events).
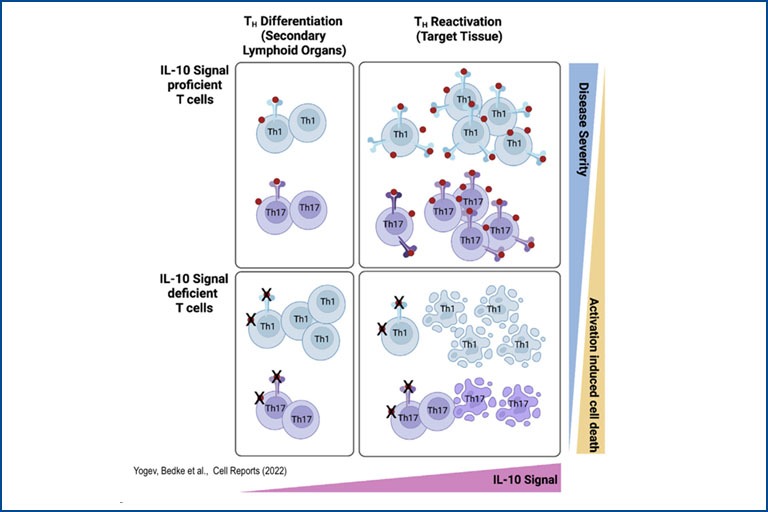
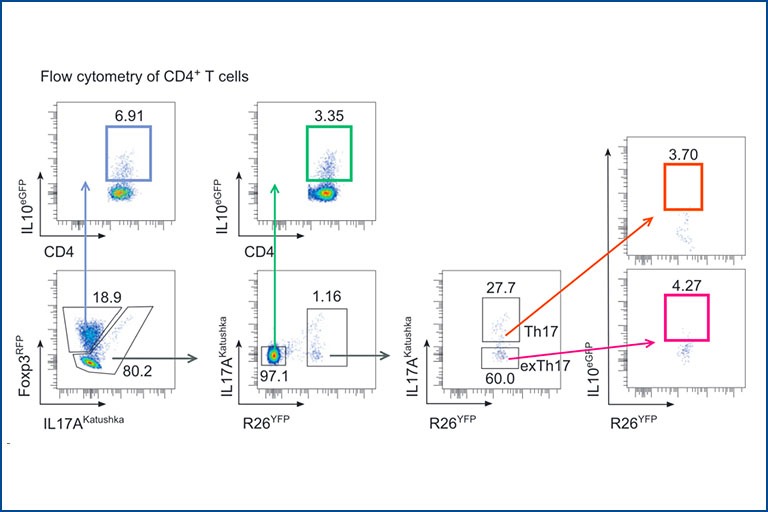
Regulatory T cells in Immune mediated inflammatory diseases
Regulatory T cells (Treg) play a pivotal role in maintaining immune homeostasis and controlling chronic inflammation and autoimmunity. In the intestine, Treg are essential for maintaining the delicate balance between the immune system and the gut microbiota, thereby preventing conditions such as inflammatory bowel disease (IBD) and promoting tolerance to beneficial gut bacteria, ensuring intestinal health. Similarly, in extra-intestinal tissues like the liver and central nervous system, Treg mitigate inflammation in chronic diseases such as primary sclerosing cholangitis (PSC) and multiple sclerosis (MS) by controlling autoreactive immune cells to prevent tissue damage and maintain organ function. Our research aims to elucidate the mechanisms by which Treg control both intra- and extra-intestinal inflammation using basic mouse models and translational approaches. We assess their regulatory function in various tissue environments, on other immune cells such as Th17 cells, and the involved signaling pathways. This knowledge is crucial for developing effective treatments for inflammatory diseases, potentially leading to improved management and cure of autoimmune and chronic inflammatory conditions.
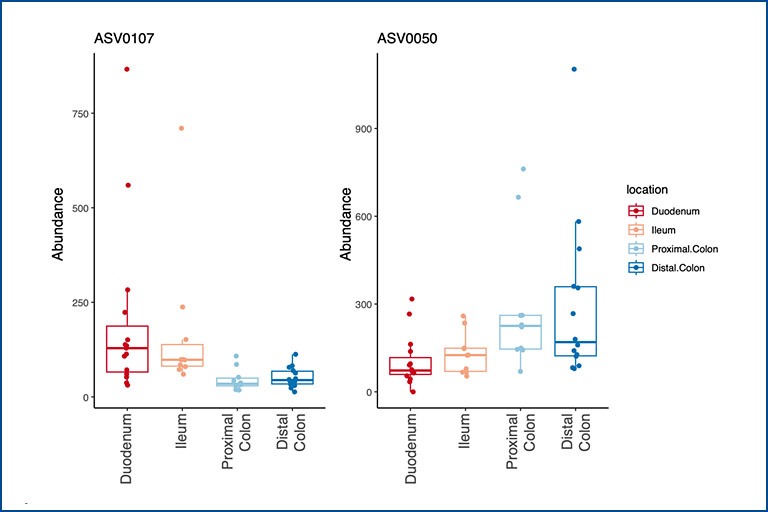
Influence of the microbiota on intestinal dendritic cells and its impact on Inflammatory Bowel Disease
Intestinal dendritic cells (DCs) play a central role in maintaining homeostasis and initiating adaptive immune responses within the gastrointestinal tract. Not only must they interact with the complex microbial milieu but at the same time prime and modulate the differentiation of T cells. So far, the study of intestinal DCs has been encumbered by their limited numbers and phenotypic overlap with other myeloid cells. Thus, the precise influence of the local microenvironment, and in particular the microbiota, on the functional and phenotypic attributes of intestinal DCs is not completely understood. Studying how intestinal DCs are influenced by their environment and how it affects their interaction with T cells will provide insights into the mechanisms involved in intestinal homeostasis and diseases such as Inflammatory Bowel Disease (IBD).
Publications 2026
Lanton, T., Eidelshtein, D., Rachmilewitz, J., Abramovitch, R., Pappo, O., Udi, S., Baraghithy, S., Tam, J., Perles, S., Williams, E., Elgavish, S., Ruppo, S., Benyamini, H., Mor, U., Elinav, E., Schmidt-Arras, D., Rehman, A., Rosenstiel, P., Giannou, A., Huber, S., Rose-John, S., Galun, E. & Axelrod, J. H., 2026, in: GASTRO HEP ADV. 5, 2, S. 100819
Abstract
Background and Aims: Interleukin-6 (IL-6) performs multiple roles in regulating metabolic pathways in both mice and man. Here, we examined the age-dependent metabolic phenotype of SGP mice – mice overexpressing sgp130, a factor that specifically blocks IL-6 trans-signaling – that were housed in distant vivaria.
Methods: Transgenic SGP mice engineered to block IL-6 trans-signaling and wild-type littermates were raised in a Jerusalem animal facility to up to 14 months of age and assessed for weight gain, body composition, and metabolic determinants of energy expenditure in young versus aged mice. Proteomic and RNA-seq analyses were performed on liver samples as a function of age and genotype.
Results: At ∼6 months of age, weight gain, body fat accumulation, hepatosteatosis, hyperglycemia, and macrophage recruitment to adipose tissue emerged and progressed with age in SGP mice maintained in the Jerusalem animal facility, but not in 3 other vivaria. IL-6/sIL-6R blockade strongly reduced signal transducer and activator of transcription 3 phosphorylation in the liver, and hepatocyte-targeted ablation of signal transducer and activator of transcription 3 recapitulated the IL-6 trans-signaling blockade phenotype. Multiomics analyses of mouse livers revealed age- and genotype-related changes in gene expression profiles attributable to bacterial byproducts. Depletion of the gut microbiota by antibiotic treatment from the age of 6 months reversed the obese phenotype in transgenic mice, confirming the crucial role of the microbiome in the phenotype. Accordingly, the microbiome of mice from the Jerusalem animal facility differed significantly from that of mice from animal facilities in Kiel and Hamburg, Germany, where the same mice did not develop a metabolic phenotype.
Conclusion: These findings reveal the crucial functions of IL-6 trans-signaling in preventing mature-onset body fat accumulation induced by certain intestinal microbiota.
Kocheise, L., Kempski, J., Tang, Y., Balcar, L., Berger, V., Tomczak, M., Gorgulho, J., Masood, R., Giehren, F., Schmidt, C., Sutter, J. P., Fruendt, T. W., Pagani, F., Wulf, S., Kött, J., Domanig, S., Giannou, A., Bedke, T., Lücke, J., Zhang, T., Zhang, S., Machicote, A., Lohse, A. W., Alunni-Fabbroni, M., Ricke, J., Khaled, N. B., Pinter, M., Scheiner, B., Huber, S., von Felden, J., Piseddu, I. & Schulze, K., 2026, in: ANN HEPATOL. 31, 1, S. 102138
Abstract
Introduction and Objectives:
Immunotherapy with atezolizumab/bevacizumab (atezo/bev) is an established first-line treatment for patients with non-resectable hepatocellular carcinoma (HCC). Despite notable successes, only a subset of patients shows treatment response, highlighting the need for biomarkers to identify those likely to benefit from this therapy.
Materials and Methods: In this biomarker study, 143 patients with atezo/bev-treated HCC were enrolled across three European centers. Baseline cytokine levels were measured using a flow cytometric multiplex bead assay. Overall survival (OS) analysis, reported as hazard ratios (HR), was conducted in an unbiased manner, with patients divided into a discovery cohort (one center, 63 patients) and a validation cohort (two centers, 80 patients).
Results: Our cohorts show typical baseline characteristics of Western HCC patients, with alcohol-related liver disease (35.0 %) and hepatitis C (21.7 %) as the main HCC etiologies. Elevated serum IL-6 (cut-off 18.22 pg/ml) was associated with poor OS in both the discovery (HR 2.6, 95 % CI 1.2–5.6, p = 0.013) and validation cohorts (HR 2.4, 95 % CI 1.3–4.4, p = 0.005). Multivariate analysis confirmed elevated IL-6 to be a significant prognostic biomarker of poor OS (HR 2.1, 95 % CI 1.1–3.9, p = 0.021) after adjusting for established risk factors.
Conclusions: We identify elevated serum IL-6 levels as prognostic biomarker in patients with advanced HCC in Western countries. Importantly, this association was independent of infection with viral hepatitis, thus extending the previously reported associations between IL-6 and treatment response in East Asian cohorts.
Publications 2025
Seeger, P., Preukschas, A. A., Lücke, J., Sabihi, M., Stringa, P., Lausada, N., Moreira, J., Dezillio, L. E. V., Serradilla, J., Moreno, A. M. A., Oliveros, F. H., Bennemann, S., Machicote, A., Zazara, D. E., Papazoglou, E. D., Mercanoglu, B., Ghadban, T., Nentwich, M. F., Nickel, F., Mann, O., Hackert, T., Gentilini, M. V., Oh, J., Oldani, G., Chartier-Plante, S., Harriman, D., Oldhafer, K. J., Huber, S., Gondolesi, G. E. & Giannou, A. D., 12.2025, in: CURR PROB SURG. 73, S. 101917
No abstract available
Preukschas, A. A., Grotelüschen, R., Nakano, R., Suguru, M., Yokota, S., Seeger, P., Schmidt, H. C., Lücke, J., Zazara, D. E., Sabihi, M., Papazoglou, E. D., Parente, A., Wassmer, C-H., Colucci, N., Lykoudis, P. M., Ziogas, I. A., Moris, D., Mercanoglu, B., Bennemann, S., Al-Harazi, A., Nentwich, M. F., Nickel, F., Mann, O., Izbicki, J. R., Hackert, T., Oh, J., Segedi, M., Bleszynski, M., Chartier-Plante, S., Harriman, D., Wagner, K. C., Oldhafer, K. J., Lohse, A. W., Arck, P., Gondolesi, G. E., Huber, S., Sayed, B. A., Vandermeulen, M., Oldani, G., Belenkov, S., Li, J., Tomuschat, C., Dust, T., Moreno, A. M. A., Giorgakis, E., Adams, M. A., Mazariegos, G., Zhang, T., Thomson, A. W. & Giannou, A. D., 12.2025, in: CURR PROB SURG. 73, S. 101920
No abstract available
Horst, L. J., Kempski, J., Walmsley, M., Huber, S. & Schramm, C., 01.10.2025, in: HEPATOLOGY. 82, 4, S. 960-984 25 S.
Abstract
Primary sclerosing cholangitis is one of the most challenging conditions in hepatology, and due to our limited understanding of its pathogenesis, no causal therapies are currently available. While it was long assumed that a minority of people with inflammatory bowel disease (IBD) also develop primary sclerosing cholangitis (PSC), which is sometimes labeled an extraintestinal manifestation of IBD, the clinical phenotype, genetic, and intestinal microbiota associations strongly argue for PSC-IBD being a distinct form of IBD, existing alongside ulcerative colitis and Crohn’s disease. In fact, the liver itself could contribute to intestinal pathology, clinically overt in 60%–80% of patients. Recent studies suggested that on a molecular level, almost all people with PSC have underlying colitis. The extent to which the liver and gut influence each other clinically and in terms of disease progression has not yet been conclusively revealed. However, while it seemed intuitive that the 2 diseases have a negative influence on each other, evidence suggests that sclerosing cholangitis can also be protective for the gut and that colitis can, in certain settings, ameliorate liver pathology. This underscores the complex pathophysiological relationships, where factors such as genetic predisposition, changes in the intestinal microbiota, altered bile acid metabolism, and immune cell migration are among the suspected contributors. PSC is an emerging disease with a significant impact on the health-related quality of life of affected people. With this review, we aim to summarize the current knowledge on the gut-liver axis in PSC-IBD, provide new perspectives on risk stratification and treatment, and identify gaps in our current knowledge. Our understanding of this complex relationship will therefore help to design clinical trials and shape the future therapy of PSC-IBD.
Letz, P., Huber, S. & Velasquez, L. N., 30.09.2025, in: SEMIN IMMUNOPATHOL. 47, 1, S. 37
Abstract
Conventional dendritic cells (cDCs) play a pivotal role in orchestrating the delicate balance between immunity and tolerance within the gastrointestinal tract by interacting with other cell types, particularly T cells. Meanwhile, the microbiota is critical for the induction and modulation of the immune system in the gut and plays a key role in the function of cDCs. So far, the study of intestinal cDCs has been encumbered by their limited numbers and phenotypic overlap with other myeloid cells. Recent advancements in single-cell sequencing technology have helped define cDCs and their subsets, while also providing valuable insights into the contribution of cDCs to Inflammatory Bowel Disease (IBD). However, the exact role of cDCs in IBD remains unclear, particularly in terms of how the microbiota influences their function in this context. In this review, we summarize the functions of cDCs in the intestine and during IBD, and the role of the microbiota in cDC biology. We also describe the current limitations in the study of cDCs and the microbiota, as well as new methods for studying DC-T cell communications in vivo, which can help increase our understanding of the function of cDCs in the intestine and develop new therapeutic strategies against IBD.
Shiri, A. M., Fard-Aghaie, M., Bedke, T., Papazoglou, E. D., Sabihi, M., Zazara, D. E., Zhang, S., Lücke, J., Seeger, P., Evers, M., Hackert, T., Oldhafer, K. J., Gondolesi, G. E., Huber, S. & Giannou, A. D., 23.09.2025, in: SCI REP-UK. 15, S. 32641
Correction to: Scientific Reports
The original version of this Article contained errors.
In the original version of this Article, Ahmad Mustafa Shiri and Mohammad Fard-Aghaie were omitted as equally contributing authors.
In addition, due to an error during figure assembly in Figure 2C, the FACS panel for the condition “steady state” CD3 + shows the same image as the condition “metastasis” CD3-. In addition, the FACS profiles in Figures 2B–E and Figure 4A, C, E are missing numbers on all axes.
Sutter, J. P., Kocheise, L., Kempski, J., Christner, M., Wichmann, D., Pinnschmidt, H., Schmiedel, S., Lohse, A. W., Huber, S. & Brehm, T. T., 06.2025, in: INFECTION. 53, 3, S. 1249-1250 2 S.
No abstract available.
Schulze, K., Rose, T. D., Adlung, L., Peschka, M., Pagani, F., Gorgulho, J., Fründt, T. W., Labgaa, I., Haber, P. K., Zimpel, C., Castven, D., Weinmann, A., Garzia-Lezana, T., Waldmann, M., Renné, T., Voß, H., Moritz, M., Orlikowski, D., Schlüter, H., Baumbach, J., Schwartz, M., Lohse, A. W., Huber, S., Sangro, B., Macias, R. I. R., Izquierdo-Sanchez, L., Banales, J. M., Wege, H., Marquardt, J. U., Villanueva, A., Pauling, J. K. & von Felden, J., 05.2025, in: JHEP REP. 7, 5, S. 101340
Abstract
Background & Aims: Actionable candidates of hepatocarcinogenesis remain elusive, and tools for early detection are suboptimal. Our aim was to demonstrate that serum metabolome profiles reflect the initiation of hepatocellular carcinoma (HCC) and enable the identification of biomarkers for early HCC detection and actionable candidates for chemoprevention.
Methods: This global cohort study included 654 patients and 801 biospecimens. Following serum metabolome profiling across the spectrum of hepatocarcinogenesis, we conducted a phase II biomarker case–control study for early HCC detection. Findings were independently validated through in silico analysis, mRNA sequencing, and proteome profiling of primary HCC and non-tumoral tissue, and in vitro experiments.
Results: Aspartic acid, glutamic acid, taurine, and hypoxanthine were differentially abundant in the serum across chronic liver disease, cirrhosis, initial HCC, and progressed HCC, independent of sex, age, and etiology. In a phase II biomarker case–control study, a blood-based metabolite signature yielded an AUC of 94% to discriminate between patients with early-stage HCC and controls with cirrhosis, including independent validation. Unsupervised biclustering (MoSBi), lipid network analysis (LINEX2), and pathway enrichment analysis confirmed alterations in amino acid-, lipid-, and nucleotide-related pathways. In tumor tissue, these pathways were significantly deregulated regarding gene and protein expression in two independent datasets, including actionable targets RRM2, GMPS, BCAT1, PYCR2, and NEU1. In vitro knockdown confirmed a functional role in proliferation and migration, as exemplified for PYCR2.
Conclusions: These findings demonstrate that serum metabolome profiling indicates deregulated metabolites and pathways during hepatocarcinogenesis. Our liquid biopsy approach accurately detects early-stage HCC outperforming currently recommended surveillance tools and facilitates identification of actionable candidates for chemoprevention.
Impact and implications: Deregulated cellular metabolism is a hallmark of cancer. In smaller studies, circulating metabolite profiles have been associated with HCC, although mainly in the context of fatty liver disease. Translation strategies for primary prevention or early detection are lacking. In this global study, we present an unsupervised landscape of the altered serum metabolome profile during hepatocarcinogenesis, independent of age, sex, and etiology. We provide a blood-based metabolite signature that accurately identifies early-stage HCC in a phase II biomarker study including independent validation. Further RRM2, GMPS, BCAT1, PYCR2, and NEU1 are identified in tumor tissue as actionable candidates for prevention. Our data provide the rationale for clinical trials testing liquid biopsy metabolome-based signatures for early HCC detection and the development of chemoprevention strategies.
Henze, L., Will, N., Lee, D., Haas, V., Casar, C., Meyer, J., Stein, S., Mangler, F., Steinmann, S., Poch, T., Krause, J., Fuss, J., Schröder, J., Kulle, A. E., Holterhus, P-M., Bonn, S., Altfeld, M., Huber, S., Lohse, A. W., Schwinge, D. & Schramm, C., 22.04.2025, in: JCI INSIGHT. 10, 8, S. e184544
Abstract
Autoimmune hepatitis (AIH) and primary biliary cholangitis (PBC) are autoimmune liver diseases with strong female predominance. They are caused by T cell–mediated injury of hepatic parenchymal cells, but the mechanisms underlying this sex bias are unknown. Here, we investigated whether testosterone contributes to T cell activation in women with PBC. Compared with sex- and age-matched healthy controls (n = 23), cisgender (cis) women with PBC (n = 24) demonstrated decreased testosterone serum levels and proinflammatory CD4+ T cell profile in peripheral blood. Testosterone suppressed the expression of TNF and IFN-γ by human CD4+ T cells in vitro. In trans men receiving gender-affirming hormone therapy (GAHT) (n= 25), testosterone affected CD4+ T cell function by inhibiting Th1 and Th17 differentiation and by supporting the differentiation into regulatory Treg. Mechanistically, we provide evidence for a direct effect of testosterone on T cells using mice with T cell–specific deletion of the cytosolic androgen receptor. Supporting a role for testosterone in autoimmune liver disease, we observed an improved disease course and profound changes in T cell states in a trans man with AIH/primary sclerosing cholangitis (PSC) variant syndrome receiving GAHT. We here report a direct effect of testosterone on CD4+ T cells that may contribute to future personalized treatment strategies.
Piecha, F., Jahn, B-V., Köntopf, J., Koop, A., Ozga, A-K., Al-Jawazneh, A., Harberts, A., Riedel, C., Buggisch, P., Benten, D., Hübener, P., Adam, G., Huber, S., Lohse, A. W., Bannas, P. & Kluwe, J., 04.2025, in: LIVER INT. 45, 4, S. e16156
Abstract
Background and aims: Portal hypertension is the main pathophysiological driver of decompensation in patients with liver cirrhosis. Epithelial cell death markers, m30 and m65, correlate with hepatic injury and predict outcomes across various stages of liver disease. We aim (i) to evaluate whether portal hypertension itself contributes to liver outcome-relevant epithelial injury, and (ii) to analyse the capacity of m30/m65 to predict outcome in patients receiving a transjugular intrahepatic portosystemic shunt (TIPS) for refractory ascites.
Methods: Sixty-six patients undergoing TIPS placement for refractory ascites and 20 patients with compensated cirrhosis as controls were prospectively enrolled in this monocentric cohort study. Epithelial cell death markers were analysed pre-TIPS, as well as 1-3 and 6-9 months post-TIPS. The capacity of baseline levels of m30/m65 in predicting six-month transplant-free survival rates was analysed by multivariable Cox proportional hazards regression.
Results: Levels of m30 and m65 were higher in patients with decompensated cirrhosis (pre-TIPS) compared with compensated cirrhosis (controls). Following correction of portal hypertension by TIPS and recompensation, both markers decreased over time, reaching levels comparable to patients with compensated cirrhosis. On multivariable analysis, pre-TIPS baseline levels of m30 and m65 were not predictive for six-month survival.
Conclusion: Correction of portal hypertension via TIPS reduces levels of epithelial cell death markers, indicating that portal hypertension is a driver of outcome-relevant, hepatic cell death in patients with decompensated cirrhosis. Baseline m30/m65 values do not affect six-month survival rates, which suggests that TIPS placement overcomes the unfavourable spontaneous prognosis otherwise indicated by elevated baseline m30/65 levels.
Nawrocki, M. & Huber, S., 04.2025, T Cell Activation: Methods and Protocols. Diercks, B-P. (Hrsg.). 1 Aufl. New York, NY: Humana Press, S. 125-132 8 S. (Methods Mol Biol; Band 2904).
Abstract
The hallmark of the adaptive immune response is its ability to adjust to the offending agents via antigen specificity and the type of immune mechanisms involved in the host defense. The cytokine milieu present in the inflammatory environment and the strength of T cell receptor (TCR) signaling control the differentiation of CD4+ T cells into specific effector or regulatory lineages. Nevertheless, CD4+ T cell differentiation is a dynamic ongoing process allowing for the plasticity of lineages, with CD4+ T cells possessing phenotypes of more than one effector lineage and transdifferentiation between effector states or from an effector into a regulatory phenotype. In this protocol, we describe how the use of reporter mice can be employed for studying cell transdifferentiation of CD4+ T cells in vitro.
Das, S., Parigi, S. M., Luo, X., Fransson, J., Kern, B. C., Okhovat, A., Diaz, O. E., Sorini, C., Czarnewski, P., Webb, A. T., Morales, R. A., Lebon, S., Monasterio, G., Castillo, F., Tripathi, K. P., He, N., Pelczar, P., Schaltenberg, N., De la Fuente, M., López-Köstner, F., Nylén, S., Larsen, H. L., Kuiper, R., Antonson, P., Hermoso, M. A., Huber, S., Biton, M., Scharaw, S., Gustafsson, J-Å., Katajisto, P. & Villablanca, E. J., 01.2025, in: NATURE. 637, 8048, S. 1198-1206 9 S.
Abstract
Uncontrolled regeneration leads to neoplastic transformation. The intestinal epithelium requires precise regulation during continuous homeostatic and damage-induced tissue renewal to prevent neoplastic transformation, suggesting that pathways unlinking tumour growth from regenerative processes must exist. Here, by mining RNA-sequencing datasets from two intestinal damage models4,5 and using pharmacological, transcriptomics and genetic tools, we identified liver X receptor (LXR) pathway activation as a tissue adaptation to damage that reciprocally regulates intestinal regeneration and tumorigenesis. Using single-cell RNA sequencing, intestinal organoids, and gain- and loss-of-function experiments, we demonstrate that LXR activation in intestinal epithelial cells induces amphiregulin (Areg), enhancing regenerative responses. This response is coordinated by the LXR-ligand-producing enzyme CYP27A1, which was upregulated in damaged intestinal crypt niches. Deletion of Cyp27a1 impaired intestinal regeneration, which was rescued by exogenous LXR agonists. Notably, in tumour models, Cyp27a1 deficiency led to increased tumour growth, whereas LXR activation elicited anti-tumour responses dependent on adaptive immunity. Consistently, human colorectal cancer specimens exhibited reduced levels of CYP27A1, LXR target genes, and B and CD8 T cell gene signatures. We therefore identify an epithelial adaptation mechanism to damage, whereby LXR functions as a rheostat, promoting tissue repair while limiting tumorigenesis.
Bugaichuk, S., Wilkens, V., Horvatits, K., Huber, S., Lohse, A. W., Kluwe, J., Pischke, S. & Fründt, T., 2025, in: ANN HEPATOL. 30, 2, S. 101926
Abstract
Introduction and objectives: Elevated liver enzymes (ELE) are a common finding in the general population, often caused by undiagnosed chronic liver disease. But little is known to what extent socioeconomic status (SES) influences the occurrence of various liver diseases.
Material and methods: Retrospective study of outpatients presenting with ELE. All patients received a structured work-up including abdominal ultrasound and serological testing. SES was assessed for patients from the Hamburg area using the social monitoring database of the Hamburg City Housing Department. SES was rated as high (SES-H), medium (SES-M), and low (SES-L).
Results: Out of n=859 patients analysed, SES was assessable for n = 310 (53%) patients: SES-H/-M/-L [n; %]: 31 (10%), 223 (72%), 56 (18%). The most prevalent liver diseases were NAFLD (n=125; 40.3%), drug-induced liver injury (n=16; 5.2%) and alcoholic liver disease (ALD, n=13; 4.2%). Prevalence of NAFLD differed significantly between SES-subgroups (SES-H/-M/-L [n; %]: 6 (19%) vs. 88 (39%) vs. 32 (55%); p= .004), the distribution of ALD was similar between the SES subgroups (1(3.2%) vs. 11 (4.9%) vs. 1 (2%); p= .55). Median body mass index (BMI) increased from SES-H to SES-VL (SES-H/-M/-L [kg/m2]: 24.4 vs. 26.2 vs. 28.6; p= .001).
Conclusions: NAFLD is the most prevalent liver disease in patients presenting with unexplained ELE, with a significantly higher occurrence in individuals from lower SES groups. Furthermore, BMI increases among patients with lower SES, highlighting the potential role of socioeconomic factors in NAFLD development. These findings underscore the need for targeted public health interventions, particularly in socioeconomically disadvantaged population.
Publications 2024
Abstract
Adeno-associated virus (AAV) is a promising in vivo gene delivery platform showing advantages in delivering therapeutic molecules to difficult or undruggable cells. However, natural AAV serotypes have insufficient transduction specificity and efficiency in kidney cells. Here, we developed an evolution-directed selection protocol for renal glomeruli and identified what we believe to be a new vector termed AAV2-GEC that specifically and efficiently targets the glomerular endothelial cells (GEC) after systemic administration and maintains robust GEC tropism in healthy and diseased rodents. AAV2-GEC-mediated delivery of IdeS, a bacterial antibody-cleaving proteinase, provided sustained clearance of kidney-bound antibodies and successfully treated antiglomerular basement membrane glomerulonephritis in mice. Taken together, this study showcases the potential of AAV as a gene delivery platform for challenging cell types. The development of AAV2-GEC and its successful application in the treatment of antibody-mediated kidney disease represents a significant step forward and opens up promising avenues for kidney medicine.
Abstract
Background: Minimal change disease and primary focal segmental glomerulosclerosis in adults, along with idiopathic nephrotic syndrome in children, are immune-mediated podocytopathies that lead to nephrotic syndrome. Autoantibodies targeting nephrin have been found in patients with minimal change disease, but their clinical and pathophysiological roles are unclear.
Methods: We conducted a multicenter study to analyze antinephrin autoantibodies in adults with glomerular diseases, including minimal change disease, focal segmental glomerulosclerosis, membranous nephropathy, IgA nephropathy, antineutrophil cytoplasmic antibody-associated glomerulonephritis, and lupus nephritis, as well as in children with idiopathic nephrotic syndrome and in controls. We also created an experimental mouse model through active immunization with recombinant murine nephrin.
Results: The study included 539 patients (357 adults and 182 children) and 117 controls. Among the adults, antinephrin autoantibodies were found in 46 of the 105 patients (44%) with minimal change disease, 7 of 74 (9%) with primary focal segmental glomerulosclerosis, and only in rare cases among the patients with other conditions. Of the 182 children with idiopathic nephrotic syndrome, 94 (52%) had detectable antinephrin autoantibodies. In the subgroup of patients with active minimal change disease or idiopathic nephrotic syndrome who were not receiving immunosuppressive treatment, the prevalence of antinephrin autoantibodies was as high as 69% and 90%, respectively. At study inclusion and during follow-up, antinephrin autoantibody levels were correlated with disease activity. Experimental immunization induced a nephrotic syndrome, a minimal change disease-like phenotype, IgG localization to the podocyte slit diaphragm, nephrin phosphorylation, and severe cytoskeletal changes in mice.
Conclusions: In this study, circulating antinephrin autoantibodies were common in patients with minimal change disease or idiopathic nephrotic syndrome and appeared to be markers of disease activity. Their binding at the slit diaphragm induced podocyte dysfunction and nephrotic syndrome, which highlights their pathophysiological significance. (Funded by Deutsche Forschungsgemeinschaft and others.).
Abstract
Objective: There is a strong clinical association between IBD and primary sclerosing cholangitis (PSC), a chronic disease of the liver characterised by biliary inflammation that leads to strictures and fibrosis. Approximately 60% – 80% of people with PSC will also develop IBD (PSC-IBD). One hypothesis explaining this association would be that PSC drives IBD. Therefore, our aim was to test this hypothesis and to decipher the underlying mechanism.
Design: Colitis severity was analysed in experimental mouse models of colitis and sclerosing cholangitis, and people with IBD and PSC-IBD. Foxp3+ Treg-cell infiltration was assessed by qPCR and flow cytometry. Microbiota profiling was carried out from faecal samples of people with IBD, PSC-IBD and mouse models recapitulating these diseases. Faecal microbiota samples collected from people with IBD and PSC-IBD were transplanted into germ-free mice followed by colitis induction.
Results: We show that sclerosing cholangitis attenuated IBD in mouse models. Mechanistically, sclerosing cholangitis causes an altered intestinal microbiota composition, which promotes Foxp3+ Treg-cell expansion, and thereby protects against IBD. Accordingly, sclerosing cholangitis promotes IBD in the absence of Foxp3+ Treg cells. Furthermore, people with PSC-IBD have an increased Foxp3+ expression in the colon and an overall milder IBD severity. Finally, by transplanting faecal microbiota into gnotobiotic mice, we showed that the intestinal microbiota of people with PSC protects against colitis.
Conclusion: This study shows that PSC attenuates IBD and provides a comprehensive insight into the mechanisms involved in this effect.
Abstract
Background & Aims: The liver is one of the organs most commonly affected by metastasis. The presence of liver metastases has been reported to be responsible for an immunosuppressive microenvironment and diminished immunotherapy efficacy. Herein, we aimed to investigate the role of IL-10 in liver metastasis and to determine how its modulation could affect the efficacy of immunotherapy in vivo.
Methods: To induce spontaneous or forced liver metastasis in mice, murine cancer cells (MC38) or colon tumor organoids were injected into the cecum or the spleen, respectively. Mice with complete and cell type-specific deletion of IL-10 and IL-10 receptor alpha were used to identify the source and the target of IL-10 during metastasis formation. Programmed death ligand 1 (PD-L1)-deficient mice were used to test the role of this checkpoint. Flow cytometry was applied to characterize the regulation of PD-L1 by IL-10.
Results: We found that Il10-deficient mice and mice treated with IL-10 receptor alpha antibodies were protected against liver metastasis formation. Furthermore, by using IL-10 reporter mice, we demonstrated that Foxp3+ regulatory T cells (Tregs) were the major cellular source of IL-10 in liver metastatic sites. Accordingly, deletion of IL-10 in Tregs, but not in myeloid cells, led to reduced liver metastasis. Mechanistically, IL-10 acted on Tregs in an autocrine manner, thereby further amplifying IL-10 production. Furthermore, IL-10 acted on myeloid cells, i.e. monocytes, and induced the upregulation of the immune checkpoint protein PD-L1. Finally, the PD-L1/PD-1 axis attenuated CD8-dependent cytotoxicity against metastatic lesions.
Conclusions: Treg-derived IL-10 upregulates PD-L1 expression in monocytes, which in turn reduces CD8+ T-cell infiltration and related antitumor immunity in the context of colorectal cancer-derived liver metastases. These findings provide the basis for future monitoring and targeting of IL-10 in colorectal cancer-derived liver metastases.
Impact and implications: Liver metastasis diminishes the effectiveness of immunotherapy and increases the mortality rate in patients with colorectal cancer. We investigated the role of IL-10 in liver metastasis formation and assessed its impact on the effectiveness of immunotherapy. Our data show that IL-10 is a pro-metastatic factor involved in liver metastasis formation and that it acts as a regulator of PD-L1. This provides the basis for future monitoring and targeting of IL-10 in colorectal cancer-derived liver metastasis.
Publications 2023
Abstract
Current therapies for Fabry disease are based on reversing intracellular accumulation of globotriaosylceramide (Gb3) by enzyme replacement therapy (ERT) or chaperone-mediated stabilization of the defective enzyme, thereby alleviating lysosomal dysfunction. However, their effect in the reversal of end-organ damage, like kidney injury and chronic kidney disease, remains unclear. In this study, ultrastructural analysis of serial human kidney biopsies showed that long-term use of ERT reduced Gb3 accumulation in podocytes but did not reverse podocyte injury. Then, a CRISPR/Cas9-mediated α-galactosidase knockout podocyte cell line confirmed ERT-mediated reversal of Gb3 accumulation without resolution of lysosomal dysfunction. Transcriptome-based connectivity mapping and SILAC-based quantitative proteomics identified α-synuclein (SNCA) accumulation as a key event mediating podocyte injury. Genetic and pharmacological inhibition of SNCA improved lysosomal structure and function in Fabry podocytes, exceeding the benefits of ERT. Together, this work reconceptualizes Fabry-associated cell injury beyond Gb3 accumulation, and introduces SNCA modulation as a potential intervention, especially for patients with Fabry nephropathy.
Abstract
Expansion microscopy physically enlarges biological specimens to achieve nanoscale resolution using diffraction-limited microscopy systems. However, optimal performance is usually reached using laser-based systems (for example, confocal microscopy), restricting its broad applicability in clinical pathology, as most centres have access only to light-emitting diode (LED)-based widefield systems. As a possible alternative, a computational method for image resolution enhancement, namely, super-resolution radial fluctuations (SRRF), has recently been developed. However, this method has not been explored in pathology specimens to date, because on its own, it does not achieve sufficient resolution for routine clinical use. Here, we report expansion-enhanced super-resolution radial fluctuations (ExSRRF), a simple, robust, scalable and accessible workflow that provides a resolution of up to 25 nm using LED-based widefield microscopy. ExSRRF enables molecular profiling of subcellular structures from archival formalin-fixed paraffin-embedded tissues in complex clinical and experimental specimens, including ischaemic, degenerative, neoplastic, genetic and immune-mediated disorders. Furthermore, as examples of its potential application to experimental and clinical pathology, we show that ExSRRF can be used to identify and quantify classical features of endoplasmic reticulum stress in the murine ischaemic kidney and diagnostic ultrastructural features in human kidney biopsies.
Abstract
Membranous nephropathy (MN) is an antibody-mediated autoimmune disease characterized by glomerular immune complexes containing complement components. However, both the initiation pathways and the pathogenic significance of complement activation in MN are poorly understood. Here, we show that components from all three complement pathways (alternative, classical and lectin) are found in renal biopsies from patients with MN. Proximity ligation assays to directly visualize complement assembly in the tissue reveal dominant activation via the classical pathway, with a close correlation to the degree of glomerular C1q-binding IgG subclasses. In an antigen-specific autoimmune mouse model of MN, glomerular damage and proteinuria are reduced in complement-deficient mice compared with wild-type littermates. Severe disease with progressive ascites, accompanied by extensive loss of the integral podocyte slit diaphragm proteins, nephrin and neph1, only occur in wild-type animals. Finally, targeted silencing of C3 using RNA interference after the onset of proteinuria significantly attenuates disease. Our study shows that, in MN, complement is primarily activated via the classical pathway and targeting complement components such as C3 may represent a promising therapeutic strategy.
Giannou, A.D., Kempski, J., Shiri, A.M., Lücke, J., Zhang, T., Zhao, L., Zazara, D.E., Cortesi, F., Riecken, K., Amezcua Vesely, M.C., et al. (2023). Tissue resident iNKT17 cells facilitate cancer cell extravasation in liver metastasis via interleukin-22. Immunity 56, 125-142.e12.
During metastasis, cancer cells invade, intravasate, enter the circulation, extravasate, and colonize target organs. Here, we examined the role of interleukin (IL)-22 in metastasis. Immune cell-derived IL-22 acts on epithelial tissues, promoting regeneration and healing upon tissue damage, but it is also associated with malignancy. Il22-deficient mice and mice treated with an IL-22 antibody were protected from colon-cancer-derived liver and lung metastasis formation, while overexpression of IL-22 promoted metastasis. Mechanistically, IL-22 acted on endothelial cells, promoting endothelial permeability and cancer cell transmigration via induction of endothelial aminopeptidase N. Multi-parameter flow cytometry and single-cell sequencing of immune cells isolated during cancer cell extravasation into the liver revealed iNKT17 cells as source of IL-22. iNKT-cell-deficient mice exhibited reduced metastases, which was reversed by injection of wild type, but not Il22-deficient, invariant natural killer T (iNKT) cells. IL-22-producing iNKT cells promoting metastasis were tissue resident, as demonstrated by parabiosis. Thus, IL-22 may present a therapeutic target for prevention of metastasis.
Previous publications
Kempski, J., Giannou, A.D., Riecken, K., Zhao, L., Steglich, B., Lücke, J., Garcia-Perez, L., Karstens, K.-F., Wöstemeier, A., Nawrocki, M., et al. (2020). Gastroenterology 159, 1417-1430.e3.
Background & Aims
Unregulated activity of interleukin (IL) 22 promotes intestinal tumorigenesis in mice. IL22 binds the antagonist IL22 subunit alpha 2 (IL22RA2, also called IL22BP). We studied whether alterations in IL22BP contribute to colorectal carcinogenesis in humans and mice.
Methods
We obtained tumor and nontumor tissues from patients with colorectal cancer (CRC) and measured levels of cytokines by quantitative polymerase chain reaction, flow cytometry, and immunohistochemistry. We measured levels of Il22bp messenger RNA in colon tissues from wild-type, Tnf–/–, Lta–/–, and Ltb–/– mice. Mice were given azoxymethane and dextran sodium sulfate to induce colitis and associated cancer or intracecal injections of MC38 tumor cells. Some mice were given inhibitors of lymphotoxin beta receptor (LTBR). Intestine tissues were analyzed by single-cell sequencing to identify cell sources of lymphotoxin. We performed immunohistochemistry analysis of colon tissue microarraysfrom patients with CRC (1475 tissue cores, contained tumor and nontumor tissues) and correlated levels of IL22BP with patient survival times.
Results
Levels of IL22BP were decreased in human colorectal tumors, compared with nontumor tissues, and correlated with levels of lymphotoxin. LTBR signaling was required for expression of IL22BP in colon tissues of mice. Wild-type mice given LTBR inhibitors had an increased tumor burden in both models, but LTBR inhibitors did not increase tumor growth in Il22bp–/– mice. Lymphotoxin directly induced expression of IL22BP in cultured human monocyte–derived dendritic cells via activation of nuclear factor κB. Reduced levels of IL22BP in colorectal tumor tissues were associated with shorter survival times of patients with CRC.
Conclusions
Lymphotoxin signaling regulates expression of IL22BP in colon; levels of IL22BP are reduced in human colorectal tumors, associated with shorter survival times. LTBR signaling regulates expression of IL22BP in colon tumors in mice and cultured human dendritic cells. Patients with colorectal tumors that express low levels of IL22BP might benefit from treatment with an IL22 antagonist.
Brockmann, L., Soukou, S., Steglich, B., Czarnewski, P., Zhao, L., Wende, S., Bedke, T., Ergen, C., Manthey, C., Agalioti, T., et al. (2018). Nat Commun 9, 5457.
Abstract
IL-10 is a prototypical anti-inflammatory cytokine, which is fundamental to the maintenance of immune homeostasis, especially in the intestine. There is an assumption that cells producing IL-10 have an immunoregulatory function. However, here we report that IL-10-producing CD4+ T cells are phenotypically and functionally heterogeneous. By combining single cell transcriptome and functional analyses, we identified a subpopulation of IL-10-producing Foxp3neg CD4+ T cells that displays regulatory activity unlike other IL-10-producing CD4+ T cells, which are unexpectedly pro-inflammatory. The combinatorial expression of co-inhibitory receptors is sufficient to discriminate IL-10-producing CD4+ T cells with regulatory function from others and to identify them across different tissues and disease models in mice and humans. These regulatory IL-10-producing Foxp3neg CD4+ T cells have a unique transcriptional program, which goes beyond the regulation of IL-10 expression. Finally, we found that patients with Inflammatory Bowel Disease demonstrate a deficiency in this specific regulatory T-cell subpopulation.
5. Pelczar, P., Witkowski, M., Perez, L.G., Kempski, J., Hammel, A.G., Brockmann, L., Kleinschmidt, D., Wende, S., Haueis, C., Bedke, T., et al. (2016). Science 354, 6.
Abstract
Intestinal inflammation can impair mucosal healing, thereby establishing a vicious cycle leading to chronic inflammatory bowel disease (IBD). However, the signaling networks driving chronic inflammation remain unclear. Here we report that CD4+ T cells isolated from patients with IBD produce high levels of interleukin-22 binding protein (IL-22BP), the endogenous inhibitor of the tissue-protective cytokine IL-22. Using mouse models, we demonstrate that IBD development requires T cell-derived IL-22BP. Lastly, intestinal CD4+ T cells isolated from IBD patients responsive to treatment with antibodies against tumor necrosis factor-α (anti-TNF-α), the most effective known IBD therapy, exhibited reduced amounts of IL-22BP expression but still expressed IL-22. Our findings suggest that anti-TNF-α therapy may act at least in part by suppressing IL-22BP and point toward a more specific potential therapy for IBD.
Team


Prof. Dr. med. Samuel Huber
Head Huber Lab
Clinic Director
Head of Molecular Gastroenterology and Immunology
E-mail address:

Team Huber Lab
Tanja Bedke, Postdoc
Franziska Bertram, Clinician Scientist
Tom Blankenburg, Technician
Marius Böttcher, Clinician Scientist
Sogol Dostiar Tabrizi, PhD student
Gemma Douilhet, Postdoc
Can Ergen-Behr, Clinician Scientist
Anastasios Giannou, Postdoc
Cathleen Haueis, Technician
Saskia Großhauser, Technician
Jan Kempski, Clinician Scientist
Philine Letz, PhD student
Beibei Liu, PhD student
Jöran Lücke, Clinician Scientist
Andres Machicote, Postdoc
Mikolaj Nawrocki, Clinician Scientist
Justus Neuendorff, MD student
Eleftherios Papazoglou, PhD student
Penelope Pelczar, Postdoc
Amanda Pidgornij, Technician
Maya Romanskyy, MD student
Babett Steglich, Postdoc
Friederike Stumme, Postdoc
Yunhe Tang, PhD student
Huu Ban Tran, MD student
Lis Velasquez, Postdoc
Sandra Wende, Technician
Siwen Zhang, PhD student