
- Head of research group
Research Profiles
Goals
Our major interest is the proteolytic processing of certain key proteins (such as the prion protein) and its relevance in health and disease, with a special focus on neurodegenerative conditions. The latter include rare and transmissible prion diseases (e.g. Creutzfeldt-Jakob disease) and (much) more frequent diseases like Alzheimer`s or Parkinson`s disease. All of these detrimental diseases pose enormous suffering and challenges to patients, relatives, caretakers, public health services and society as a whole, and no causative –and hence really beneficial– treatments exist today. However, these diseases also share critical mechanistic processes leading to pathology which will likely be relevant for common therapeutic approaches in the future. Our strategy is to employ evolutionary conserved endogenous enzymatic cleavages (i.e., fragmentation of proteins by ‘molecular scissors‘ occuring in all of our bodies at all times). Many of those cleavages and their resulting fragments seem to play (neuro)protective roles. In other words: we try to take advantage of physiological processes already provided by nature. To this end, we constantly gain deeper mechanistic knowledge at the molecular, cellular, tissue and organismal level using various model systems and methodologies. We also develop and share new research tools (such as cleavage site-specific antibodies) enabling and accelerating critical mechanistic insight ultimately (and urgently) required for novel therapeutic perspectives. Our interdisciplinary and translational focus is also achieved by close and fruitful national and international collaborations with renowned experts in respective fields.
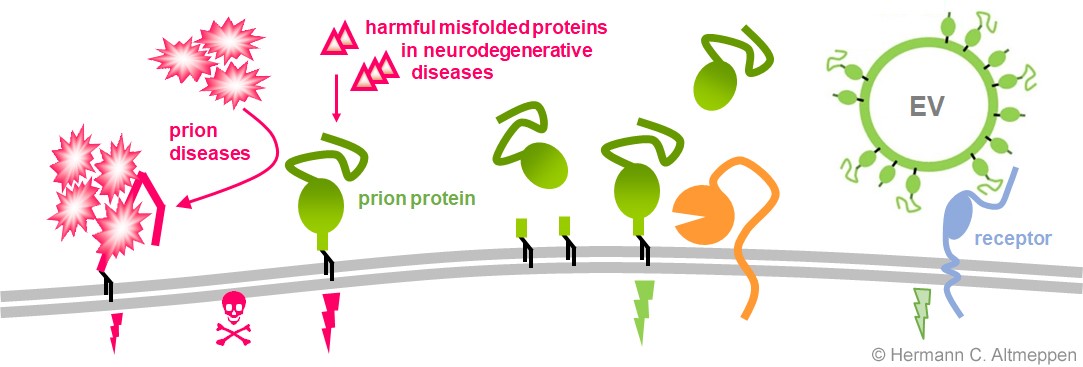
The prion protein (green) is located at the surface of neurons and many other cell types, where it exerts its physiological functions (green thunderbolt). In prion diseases (on the left) the protein is forced to misfold (i.e., changing ist three-dimensional structure) thereby loosing its functions and starting to build aggregates in a progressive process ultimately leading to death. The prion protein also acts as a cellular receptor for other toxic proteins associated with currently incurable brain diseases such as Alzheimer`s or Parkinson`s disease. Certain proteases (or „molecular scissors“, orange) in our bodies constantly cleave the prion protein at specific sites. As a consequence, levels of the protein are reduced at the cellular surface and shorter fragments of the protein are released into the extracellular space, where they may hold intrinsic biological functions and exert protective effects. Lastly, the prion protein is also released from cells in association with extracellular vesicles (EV) and may play important roles on these relevant structures in intercellular communication in health and disease.
Current projects
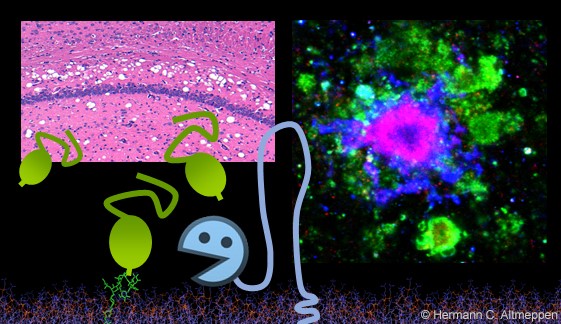
The ADAM10-mediated shedding of the prion protein – Giving a little to gain a lot?
The prion protein (PrP) is a key player in neurodegenerative diseases. Exposed on the surface of neurons it acts as a substrate for detrimental and progressive misfolding in prion diseases and as a toxicity-mediating receptor for harmful protein conformers in other neurodegenerative diseases. In contrast, release of PrP by the metalloprotease ADAM10 as well as cleavage by other enzymes reduces PrP levels at the plasma membrane and generates soluble fragments, which seem to play protective roles in neurodegenerative diseases. We are studying the biological effects of these cleavages and the resulting fragments and how these could be employed for therapy.
Selected publications
- Ligands binding to the prion protein induce its proteolytic release with therapeutic potential in neurodegenerative proteinopathies.
Linsenmeier L§, Mohammadi B§, Shafiq M, Frontzek K, Bär J, Shrivastava A, Damme M., Schwarz A, Da Vela S, Massignan T, Jung S, Correia A, Schmitz M, Puig B, Hornemann S, Zerr I, Tatzelt J, Biasini E, Saftig P, Schweizer M, Svergun D, Amin L, Mazzola F, Varani L, Thapa S, Gilch S, Schätzl H, Harris D, Triller A, Mikhaylova M, Aguzzi A, Altmeppen HC§*, Glatzel M*. (in press) Science Advances [§equal contribution; *corresponding authors]
DOI: 10.1101/2021.04.19.440495 - GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice.
Puig B, Altmeppen HC et al. (2019) PLoS Pathogens
DOI: 10.1371/journal.ppat.1007520 - Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein.
Linsenmeier L, Mohammadi B, Wetzel S, Puig B, Jackson W, Hartmann A, Uchiyama K, Sakaguchi S, Endres K, Tatzelt J, Saftig P, Glatzel M*, Altmeppen HC*. (2018) Molecular Neurodegeneration [*corresponding authors]
DOI: 10.1186/s13024-018-0248-6 - The sheddase ADAM10 is a potent modulator of prion disease.
Altmeppen HC et al. (2015) eLife
DOI: 10.7554/eLife.04260 - Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo.
Altmeppen HC et al. (2011) Molecular Neurodegeneration
DOI: 10.1186/1750-1326-6-36

Other aspects of enzymatic cleavages in PrP biology and/or neurodegenerative diseases
In addition to the aspects mentioned above, we are interested in biological functions of the prion protein and its released forms. Also, we investigate whether detection of specific PrP fragments can tell us something about disease states in neurodegeneration, cancer and other pathological conditions. To this end, we investigate the potential of such fragments as meaningful biomarkers for a better and earlier molecular diagnosis.
Together with collaborators we are also investigating the proteolytic processing and molecular involvement of other proteins critically associated with diseases of the brain and beyond.
Selected publications
- Transgenic Overexpression of the Disordered Prion Protein N1 Fragment in Mice Does Not Protect Against Neurodegenerative Diseases Due to Impaired ER Translocation.
Mohammadi B, Linsenmeier L, Shafiq M, Puig B, Galliciotti G, Giudici C, Willem M, Eden T, Koch-Nolte F, Lin YH, Tatzelt J, Glatzel M, Altmeppen HC. (2020) Molecular Neurobiology
DOI: 10.1007/s12035-020-01917-2 - Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease.
Sevillano AM, Aguilar-Calvo P, Kurt TD, Lawrence JA, Soldau K, Nam TH, Schumann T, Pizzo DP, Nyström S, Choudhury B, Altmeppen HC et al. (2020) Journal of Clinical Investigation
DOI: 10.1172/JCI131564 - In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15.
Seipold L, Altmeppen HC et al. (2018) Cellular and Molecular Life Science
DOI: 10.1007/s00018-018-2791-2 - Generation of aggregation prone N-terminally truncated amyloid β peptides by meprin β depends on the sequence specificity at the cleavage site.
Schönherr C, Schönherr C, Bien J, Isbert S, Wichert R, Prox J, Altmeppen HC et al. (2016) Molecular Neurodegeneration
DOI: 10.1186/s13024-016-0084-5 - High molecular mass assemblies of amyloid-β oligomers bind prion protein in patients with Alzheimer's disease.
Dohler F, Sepulveda-Falla D, Krasemann S, Altmeppen HC et al. (2014) Brain
DOI: 10.1093/brain/awt375
The prion protein on extracellular vesicles – functional and disease-associated aspects
The prion protein can also be released via extracellular vesicles (EV). The latter are membrane-surrounded particles shed by nearly all cell types in the body. EVs are increasingly recognized and intensively studied as important means of communication between cells with relevance in a multitude of physiological and pathological processes. They also represent a vehicle for future therapies. However, a deeper understanding of key processes (e.g. uptake and processing of EVs by recipient cells) is urgently required.
Notably, the prion protein is highly abundant on EVs, where it may exert important regulatory functions.
Selected publications
- Brain-Derived Extracellular Vesicles in Health and Disease: A Methodological Perspective.
Brenna S, Krisp C, Altmeppen HC et al. (2021) International Journal of Molecular Sciences
DOI: 10.3390/ijms22031365 - Characterization of brain-derived extracellular vesicles reveals changes in cellular origin after stroke and enrichment of the prion protein with a potential role in cellular uptake.
Brenna S, Altmeppen HC et al. (2020) Journal of Extracellular Vesicles
DOI: 10.1080/20013078.2020.1809065 - Muskelin Coordinates PrPC Lysosome versus Exosome Targeting and Impacts Prion Disease Progression.
Heisler FF, Pechmann Y, Wieser I, Altmeppen HC et al. (2018) Neuron
DOI: 10.1016/j.neuron.2018.08.010 - Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein.
Linsenmeier L, Mohammadi B, Wetzel S, Puig B, Jackson WS, Hartmann A, Uchiyama K, Sakaguchi S, Endres K, Tatzelt J, Saftig P, Glatzel M*, Altmeppen HC*. (2018) Molecular Neurodegeneration [*corresponding authors]
DOI: 10.1186/s13024-018-0248-6 - Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity.
Falker C, Hartmann A, Guett I, Dohler F, Altmeppen HC et al. (2016) Journal of Neurochemistry
DOI: 10.1111/jnc.13514
Funding Acknowledgement and Donations
We gratefully acknowledge financial support of our past and current research by the
Creutzfeldt-Jakob Disease Foundation, Inc.
; the
Alzheimer Forschung Initiative e.V.
(AFI); and the
Werner-Otto-Stiftung
.
Research is in a constant state of flux. Solving one scientific problem most certainly raises several new questions and challenges. If you want to support our current and future projects with your gracious donation (regardless of the amount – any support will help us achieving our scientific goals) you can do so with the following information, by scanning the QR code or by clicking here . Your support is very much appreciated!

DONATION ACCOUNT | SPENDENKONTO
Reference/Verwendungszweck: 1345/001
Recipient/Empfänger: UKE gemeinnützige GmbH
Bank: Hamburger Sparkasse
IBAN: DE 54 200 50 550 1234 363636
BIC: HASPDEHHXXX